Last updated: 14 Aug 25 19:11:11 (Asia/Shanghai)
药理学
This tutorial is powered by Bensz/黄伟斌
药物代谢动力学与药物效应动力学
一些基本概念。
考题
- 【例1】经肠道消化吸收的药物经过肝脏后药物浓度明显下降的原因是 A. 生物转化 B. 重吸收 C. 首过消除 D. 首剂效应 E. 肠肝循环
- 【例2】可引起首过消除的主要给药途径是 A. 吸入给药 B. 舌下给药 C. 口服给药 D. 直肠给药 E. 皮下注射
- 【例3】引起药物首过消除最主要的器官是 A. 肝 B. 肾 C. 肺 D. 肠黏膜 E. 门静脉
- 【例4】不影响药物在体内分布的因素是 A. 药物的脂溶度 B. 药物的 pKa C. 给药剂量 D. 血脑屏障 E. 器官和组织的血流量
- ✨【例5】某弱酸性药物的 pKa 是 3.4,在血浆中的解离百分率约为 A. 1% B. 10% C. 90% D. 99% E. 99.99% 解: 用 Henderson–Hasselbalch:pH = pKa + log([A−]/[HA]),得 [A−]/[HA] = 10^(pH−pKa)。 取血浆 pH ≈ 7.4,pH − pKa = 7.4 − 3.4 = 4.0,故 [A−]/[HA] = 10^4 = 10,000。 解离百分比 = [A−]/([A−]+[HA]) = 10,000/(10,000+1) ≈ 0.9999 = 99.99%。
- 【例6】按一级动力学消除的药物特点为 A. 药物的半衰期与剂量有关 B. 为绝大多数药物的消除方式 C. 单位时间内实际消除的药量不变 D. 单位时间内实际消除的药量递增 E. 体内药物经 2~3 个 t1/2 后可基本清除干净
- 【例7】一级消除动力学的特点是 A. 药物的半衰期不是恒定值 B. 为少数药物的消除方式 C. 单位时间内实际消除的药量随时间递减 D. 为一种恒速消除动力学 E. 其消除速度与初始血药浓度高低有关
- ✨【例8】以一级动力学消除的某药物,其半衰期 t1/2 为 8 小时,若按照恒定剂量每隔一个半衰期给药一次,达到稳态血药浓度所需的时间为 A. 10 小时 B. 20 小时 C. 30 小时 D. 40 小时 E. 50 小时
- 【例9】用药的间隔时间主要取决于 A. 药物与血浆蛋白的结合率 B. 药物的吸收速度 C. 药物的排泄速度 D. 药物的消除速度 E. 药物的分布速度
- 【例10】化学药品 A 适应证为原发性高血压,现拟用 B 药进行人体生物等效性研究,口服给药剂量均为 0.5 mg,A 药与 B 药的血药浓度—时间曲线下面积(AUC)分别为 27.2 ng·h/ml 和 23.3 ng·h/ml。下面关于 A 药与 B 药生物利用度的描述,正确的是 A. A 药与 B 药不具有生物等效性 B. A 药的绝对生物利用度是 54% C. B 药的绝对生物利用度是 46% D. B 药对 A 药的相对生物利用度为 86% E. A 药对 B 药的相对生物利用度为 86%
- 【例11】药物的副反应是 A. 难以避免的 B. 较严重的药物不良反应 C. 剂量过大时产生的不良反应 D. 药物作用选择性 E. 与药物治疗目的无关的效应
- 【例12】药物副作用为 A. 一般都很严重 B. 发生在大剂量情况下 C. 是可以避免的 D. 发生在治疗剂量下 E. 产生原因与药物作用的选择性低有关
- 【例13】停药后血浆中药物浓度降至阈浓度以下仍显现的药理作用称为 A. 耐受性 B. 后遗效应 C. 特异质反应 D. 副作用 E. 停药反应
- 【例14】治疗指数为 A. 比值越大就越安全 B. ED50/LD50 C. ED50/TD50 D. 比值越大,药物毒性越大 E. LD50/ED50
- 【例15】受体阻断药的特点是 A. 对受体无亲和力,无内在活性 B. 对受体无亲和力,有内在活性 C. 对受体有亲和力,无内在活性 D. 对受体有亲和力,有内在活性 E. 效应强度与其对受体的亲和力无关
基础
吸收
- 首过消除最常见的给药途径是口服给药。
- 首过消除最主要的器官是肝脏,肺和肠壁细胞也可成为首过消除的器官。
- 舌下给药后经颊黏膜吸收,可避免在肝脏迅速代谢,可在很大程度上避免肝脏的首过消除。、
- 直肠给药有 50%的药物可经下痔静脉至下腔静脉,避开肝脏,可部分避免肝脏的首过消除。
| 给药途径 | 吸收/分布特点 | 优点 | 缺点/注意点 |
|---|---|---|---|
| 消化道(口服) | 药物经胃肠道吸收,进入门静脉后经过肝脏代谢;存在显著的首过消除(首过代谢)。 | 给药方便、顺从性高,适合长期维持疗法。 | 首过消除高时生物利用度低,需增加剂量→代谢产物增多可能致毒。某些药物对胃肠刺激或不可口服者不适用。 |
| 舌下/口腔黏膜 | 药物经口腔黏膜直接进入全身循环,能显著避免肝首过效应(如硝酸甘油)。 | 起效快,可避免或减少首过消除,适用于需迅速起效的药物。 | 仅限对黏膜有良好渗透性且口腔不吞咽的制剂;口腔PH/血流影响吸收。 |
| 直肠给药 | 部分经直肠下部静脉回流可绕过肝门循环,部分药物仍被吸收并可发生首过代谢;也可用于局部作用。 | 可部分避免肝首过效应,适用于不能口服或需局部直肠给药的情况。 | 吸收不稳定,受给药部位与排便影响;局部刺激或患者依从性问题。 |
| 注射给药(静脉/肌内/皮下/动脉) | 静脉注射直接入全身循环,无吸收相;肌内/皮下通过毛细血管扩散吸收;动脉用于靶向输注。 | 起效快、剂量可控(尤其静脉),适用于急救或无法经口给药者;肌内皮下吸收较口服快。 | 侵袭性高、感染与并发症风险;部分给药(肌内/皮下)受血流影响;动脉注射具较大风险,仅在特定情形下使用。 |
| 呼吸道吸入 | 药物直接到达肺部或气道;肺可在到达全身循环前对药物有排泄/代谢,构成肺的局部首过消除。 | 可实现局部高浓度(气道/肺),起效快,系统副作用较小(若局部滞留高)。 | 吸入颗粒大小、给药方式与肺清除影响疗效;部分药物会被肺代谢,影响生物利用度。 |
| 局部用药(皮肤/眼/鼻/咽/阴道/经皮) | 主要在给药部位发挥局部作用;某些经皮或穿透性强药物可系统吸收并进入循环以维持血药浓度(如贴剂)。 | 针对局部病灶,减少全身副作用;经皮缓释贴剂可维持稳定血浓度,便于长期给药(如硝酸甘油、硝苯地平、芬太尼贴片)。 | 穿透性、皮肤完整性与给药面影响吸收;局部刺激或致敏风险;需注意系统吸收导致的全身效应。 |
分布
- 药物在体内各组织分布速度,主要取决于组织器官血流量和药物与血浆蛋白、组织细胞的结合能力; 此外,细胞膜转运体的数量和功能状态、体液 pH、生理屏障作用以及药物的分子量、化学结构、脂溶性、pKa、极性、微粒制剂的粒径等均能影啊药物的体内分布。
| 项目 | 机制 / 要点 | 临床意义 |
|---|---|---|
| 血浆蛋白结合率 | 大多数药物在血浆中有游离型与结合型并存。 结合位点与类型: • 弱酸性药物:主要与白蛋白结合。 • 弱碱性药物:主要与α1-酸性糖蛋白结合。 • 强脂溶性药物:主要与脂蛋白结合。 结合型药物不能跨膜,起到暂时贮存作用;结合特异性低,可发生竞争性置换。 | • 游离药物为有效成分,蛋白结合改变影响药效与毒性。 • 竞争性置换(合并用药)可增加游离浓度,需注意剂量与毒性。 • 低白蛋白(肝病、营养不良)可增游离药物量,调整剂量。 |
| 血脑屏障 & 胎盘屏障 | 血脑屏障(BBB):脑毛细血管内皮细胞紧密连接,周围被星形胶质细胞包绕,主要允许高脂溶性小分子通过被动扩散,阻滞大分子、水溶性或离子化药物;炎症(如脑膜炎)可使通透性增加。 胎盘屏障:胎盘绒毛与母血窦间无显著阻滞,多数组药物可穿透胎盘,胎儿血药浓度常接近母体血浆浓度。 | • 药物能否进入中枢取决于脂溶性、分子量与屏障完整性;感染时中枢用药选择会不同。 • 妊娠期慎用有致畸或胎毒药物(绝大多数药物可透过胎盘)。 |
| 体液 pH 与药物解离(酸碱捕获) | 细胞内液 pH ≈ 7.0,细胞外液 pH ≈ 7.4。弱酸/弱碱的电离受 pH 影响(Henderson–Hasselbalch 原理): • 弱酸药物在较碱性的外液更易解离(电离↑)。 • 弱碱药物在较酸性的环境更易解离。 解离型药物难以穿膜,非电离型易通过膜,导致不同室室间分布(酸化或碱化会促使药物“向另一侧滞留”)。 | • 可利用酸化或碱化改变药物/毒物在体内分布(如尿液pH调节增加排泄)。 • 酸碱状态改变(呼吸性/代谢性酸碱失衡)会影响药物分布与疗效/毒性。 |
代谢
| 项目 | 定义 | 要点 |
|---|---|---|
| 药物代谢酶 | 催化药物代谢反应的酶 | 大多数药物代谢需酶参与;肝脏为主要代谢器官(酶种类多、含量高);代谢后多数药物药理活性或毒性降低,少数增强;需代谢生成活性形式者称为前药(prodrug)。 |
| 药酶诱导药 | 能↑药物代谢酶活性,导致药物代谢加快的药物 | 诱导可引起血药浓度下降、疗效减弱或需加大剂量;对前药可增强活性生成;诱导具有时间滞后性(通常数天至数周)且可持续一段时间。 |
| 药酶抑制药 | 能↓药物代谢酶活性,导致药物代谢减慢的药物 | 抑制可导致血药浓度升高、毒性增加或需减量;对前药可减少活性生成;抑制作用通常起效较快(数小时至数天)。 |
排泄
| 项目 | 要点 | 临床意义 / 备注 |
|---|---|---|
| 定义 | 药物以原形或代谢产物通过体外通路排出的过程。 | 影响给药频率与药物清除 |
| 主要途径 | 肾排泄(主要) | 多数亲水性代谢产物主要经尿液排出 |
| 胆汁/粪便排泄(次要) | 大分子或有亲脂性且经肝代谢的药物及结合产物 | |
| 肺、汗、乳汁等 | 挥发性药物随呼气排出;少量经汗液、乳汁排出(哺乳期注意剂量与禁忌)。 | |
| 肾脏排泄 机制 | 肾小球滤过 | 无蛋白结合的低分子药物自由滤过;受血流与肾功能影响(CrCl↓ → 清除↓)。 |
| 肾小管分泌 | 有特定转运体(有机阴/阳离子转运体)介导,易发生药物相互作用(转运体竞争或抑制)。 | |
| 肾小管重吸收 | 非极性或未电离药物可被再吸收;尿液pH改变可改变重吸收(酸化或碱化尿液用于促进排泄)。 | |
| 肠肝循环 | 经肝代谢生成亲水性结合产物分泌入胆汁,进入肠腔后可被肠上皮吸收并回到肝脏,形成循环。 | 可显著延长血浆半衰期与药效持续时间;抑制肠菌β-葡萄糖苷酶或切除胆汁流可减少此循环 |
| 临床应用要点 | 评估给药方案需考虑:肾功能、肝胆排泄、尿液pH、药物与蛋白结合、转运体与酶相互作用、哺乳期用药风险。 | 肾功能减退需调整剂量;肠肝循环存在时停药后效应可延长 |
药物消除动力学
概述
- 体内药物按瞬时血药浓度以恒定的百分比消除,但单位时间内实际消除的药量随时间递减。
- 药物消除半衰期恒定,与剂量或药物浓度无关。
- 绝大多数药物都按一级动力学消除,这些药物在体内经过5个t1/2后,可基本消除干净。
- ✨每隔一个t1/2给药一次,则体内药量(或血药浓度)可逐渐累积,经过5个t1/2后,消除速度和给药速度相等,达到稳态。
| 项目 | 一级消除动力学 (First‑order) | 零级消除动力学 (Zero‑order) |
|---|---|---|
| 别称 | 线性消除过程 | 非线性(恒速)消除过程 |
| 定义 | 药物单位时间内被消除的**比例**恒定,消除速率与血浆浓度成正比。 浓度越高,每单位时间消除的量越多;半衰期为常数(与浓度相关性低) | 药物以**恒定量**/单位时间被消除,消除速率与血浆浓度无关(在该浓度范围内)。 单位时间内消除的量不随浓度改变;半衰期随浓度改变 |
| 时间-浓度曲线(线性坐标) | 在常规坐标图上呈指数下降(向下弯曲) | 在常规坐标图上呈近似直线(以恒定斜率下降) |
| 半对数坐标(ln或log浓度 vs 时间) | 呈直线,斜率 = -k(消除速率常数) → 方便计算半衰期 | 在低浓度段可呈弯曲(非线性),整体不作直线 |
| 数学表达式 | dC/dt = -k·C → C(t)=C0·e^{-kt} 半衰期 t1/2 = 0.693/k(常数) | dC/dt = -k0 (k0 为恒定消除速率,单位量/时间)→ C(t)=C0 - k0·t(直到被完全清除或转为其他动力学) 半衰期随 C0 变化 |
| 清除率(Clearance)与临床含义 | 系统性清除率(Cl)常被视为常数: Cl = k·Vd。给药剂量与浓度成比例,剂量调整按目标浓度线性推算可行。 | 表现为消除通量限制(例如代谢酶/吸收饱和);剂量增加不导致成比例浓度下降,易出现累积与毒性,需按速率(k0)或监测浓度调整给药。 |
| 常见实例 | 多数小分子药物在治疗范围内为一级消除(例如多数抗生素、β‑阻滞剂在常规剂量下) | 高剂量或饱和代谢时呈零级(如酒精(乙醇)、大剂量阿司匹林或某些解毒药在饱和时) |
| 临床评价与给药原则 | 用药监测(TDM)常可用常数半衰期设计给药间隔与维持剂量;负荷剂量按 Vd 计算。 | 需警惕用量依赖性毒性与非线性累积,遇饱和代谢应改用分次小剂量或降速率给药并加强监测。 |
单次给药的一级消除
设瞬时注入初始药量为 (A_0),一级消除常数为 (k)。药量随时间 (t) 的变化为指数衰减:
A(t)=A_0 e^{-k t}半衰期 (t_{1/2}) 与 (k) 的关系:
k=\frac{\ln 2}{t_{1/2}}经过 (n) 个半衰期(即 (t=n t_{1/2}))后剩余药量为:
A(n t_{1/2})=A_0 e^{-k n t_{1/2}}=A_0\left(e^{-\ln 2}\right)^n=A_0\left(\tfrac{1}{2}\right)^n特别地,取 (n=5):
A(5 t_{1/2})=A_0\left(\tfrac{1}{2}\right)^5=\frac{A_0}{32}\approx 0.03125\,A_0\quad(\text{约 }3.125\%)因此常说“经过约 5 个半衰期可认为已基本清除”。
累积与稳态
设给药间隔 (\tau = t_{1/2})。在时刻 (0,\tau,2\tau,\dots) 各给入一次瞬时剂量 (D)。记第 (n) 次给药后(立即给药、注入后瞬时的)体内总药量为 (A_n^+)((n) 从 0 开始,(A_0^+=D))。递推关系为:
A_n^+ = A_{n-1}^+ e^{-k\tau} + D,\qquad A_0^+=D解该等比递推(几何级数和)得到:
A_n^+ = D\sum_{i=0}^{n} \left(e^{-k\tau}\right)^i
=D\frac{1-e^{-k\tau\,(n+1)}}{1-e^{-k\tau}}令 (n\to\infty)(长期多次给药),收敛到稳态峰值 (A_{\mathrm{ss}}^+):
A_{\mathrm{ss}}^+=\lim_{n\to\infty}A_n^+ = \frac{D}{1-e^{-k\tau}}对应的稳态给药前(即在下一次给药前、刚好衰减了间隔 (\tau))的谷值:
A_{\mathrm{ss}}^- = A_{\mathrm{ss}}^+ e^{-k\tau} = \frac{D e^{-k\tau}}{1-e^{-k\tau}}当 (\tau=t_{1/2}) 时,(e{-k\tau}=e{-\ln 2}=\tfrac{1}{2})。代入上式:
- 第 (n) 次给药后(峰):
A_n^+ = D\frac{1-(1/2)^{\,n+1}}{1-1/2}=2D\bigl(1-(1/2)^{\,n+1}\bigr) - 稳态峰值:
A_{\mathrm{ss}}^+=2D - 稳态谷值:这说明在 (\tau=t_{1/2}) 情形下,稳态时“峰–谷”分别为 (2D) 与 (D),且每个间隔内消除掉的量等于一次剂量 (D),即长期时给药速率 = 消除速率(达到稳态)。
A_{\mathrm{ss}}^-=A_{\mathrm{ss}}^+\cdot\frac{1}{2}=D
稳态进程的占比(峰值相对于稳态峰的比例)为:
\frac{A_n^+}{A_{\mathrm{ss}}^+}=1-(1/2)^{\,n+1}若以半衰期个数 (N) 表示经过的总时间 (t=N t_{1/2}),则达到稳态的分数(通用形式):
\text{fraction of steady state} = 1-e^{-k t} = 1-\left(\tfrac{1}{2}\right)^{t/t_{1/2}} = 1-\left(\tfrac{1}{2}\right)^N代入 (N=5):
1-\left(\tfrac{1}{2}\right)^5 = 1-\tfrac{1}{32} = \tfrac{31}{32} \approx 0.96875\quad(\text{约 }96.875\%)因此常用“约 4–5 个半衰期可达到稳态(5 个半衰期约为 97% 的稳态)”。
药物代谢动力学参数
| 项目 | 要点 | 临床意义 / 计算 |
|---|---|---|
| 半衰期 (t1/2) | 定义:血浆药物浓度下降一半所需时间。 动力学: 一级动力学:t1/2为常数,不受初始浓度或剂量影响; 零级动力学:t1/2随浓度/剂量增加而延长(与初始浓度成正比)。 | 给药间隔通常取约一个半衰期;t1/2反映体内消除速度,决定稳态时间与剂量调整频率。 |
| 生物利用度 (F) | 定义:血管外给药后进入全身循环的药物相对量与速度。 口服F通常<100%,主要原因:吸收不完全与首过效应(肠壁、门静脉、肝脏代谢)。 分类:绝对生物利用度(AUC口服 / AUC静脉 × 100%);相对生物利用度(不同制剂间AUC比较)。 | 评价制剂等效性与给药途径选择;AUC用于定量比较:F_abs = (AUC_po / AUC_iv) × (Dose_iv / Dose_po). |
| 稳态血浆浓度 (Css) | 定义:多次给药时,单位时间进入体内的药量等于单位时间被消除的药量所达成的恒定浓度。 规律:达到稳态所需时间仅取决于药物的消除半衰期。 | 一般在4–5个t1/2可达到约94%–97%的稳态浓度。 剂量或给药间隔不变时,延长给药次数可达到稳态但不能提前达到。 |
| 负荷剂量 (Loading dose) | 目的:首次给予较大剂量以快速达到目标稳态浓度,随后用维持剂量维持。 原理:维持剂量决定稳态浓度,负荷剂量用于瞬时建立血药量。 | 口服间歇给药(间隔约1个t1/2)常采用首剂加倍;持续静脉输注时负荷剂量≈1.44×每小时维持输注量(或按 Vd×Css / F 计算)。 计算示例: Loading dose = (Vd × desired Css) / F。 |
药物效应动力学
- 药物基本作用
| 主题 | 定义 / 要点 | 临床意义 |
|---|---|---|
| 特异性(Specificity) | 指药物与某一生物分子(受体、酶等)之间的化学/结合专一性;物质基础为药物的化学结构。例:阿托品特异性阻断毒蕈碱型胆碱受体(M受体)。 | 特异性高意味着靶向性好、机制明确,但不必然等于临床选择性高;用于设计靶向治疗与预测机制相关不良反应。 |
| 选择性(Selectivity) | 指药物在体内表现出的对不同功能或器官影响的偏好程度:影响多种功能者为选择性低,仅影响单一功能者为选择性高。 特异性与选择性可能不一致(化学上的专一性高,临床表现上仍可有非靶效应)。 | 选择性高的药物通常毒副作用少、针对性强;临床用药需同时考虑剂量、分布与代谢对选择性的影响。 |
| 对因治疗(Etiologic / Causal therapy) | 目的在于消除原发致病因子,从而根治疾病。示例:抗生素杀灭致病菌、抗病毒药物抑制病毒复制。 | 治疗针对病因,可望治愈或长期控制;需明确病因和病原学诊断,存在耐药或不可逆损伤等限制。 |
| 对症治疗(Symptomatic therapy) | 目的在于缓解或控制症状而非根除病因。适用于病因不明或暂时无法根治的情况。例如止痛、退热、利尿、止吐等。 | 是临床必需手段,可改善病人生活质量与并发症预防;应与对因治疗配合,避免长期掩盖病因导致延误诊治。 |
- 不良反应
| 反应类型 | 定义 / 机制 | 临床特点与处理 |
|---|---|---|
| 副反应(副作用) | 因药物选择性低导致作用波及非治疗靶器官;为药物在治疗剂量下的固有不良效应。 | 大多可预见且较轻:如阿托品解痉时伴口干、心悸、便秘。处理:监测、减量或改用更选择性药物, symptomatic 支持。 |
| 毒性反应 | 因剂量过大或体内蓄积造成的有害反应,通常更严重且可被预见;分急性和慢性两类。 | 急性:多损害循环、呼吸、神经系统。慢性:多损害肝、肾、骨髓、内分泌,可出现致癌、致畸、致突变。处理:立即停药/解毒、支持治疗,调整剂量并避免蓄积。 |
| 后遗效应(残留效应) | 停药后血药浓度已降至最小有效浓度以下仍存在的药理效应(药物动力学/动力学延迟)。 | 例:巴比妥类次晨乏力、困倦。处理:预先告知、调整给药时间或选择半衰期更短的药物。 |
| 停药反应(反跳现象) | 长期用药后突然停药导致原有疾病或症状加重(生理/代偿性改变或受体调节所致)。 | 例:长期可乐定停药后血压回升。处理:逐步减量、替代或对症支持,必要时短期恢复给药。 |
| 变态反应(超敏反应) | 非肽类药物或其代谢物作为半抗原与机体蛋白结合,引起免疫敏化(约10天)后发生的免疫学反应;与剂量无关,与药物原有药理作用无关。 | 常见于过敏体质,范围从轻微皮疹到休克或造血/器官损害。处理:停药为主,抗组胺、糖皮质激素、支持治疗;再次用药可能复发,皮试有假阴/假阳性。 |
| 特异质反应(个体特异敏感) | 少数患者因先天遗传或代谢酶缺陷对某药异常敏感,反应与药物固有药理作用一致或成比例,与免疫无关。 | 例:琥珀酰胆碱致延长肌松由先天性血浆胆碱酯酶缺乏引起。处理:识别高危、避用或准备相应支持(如呼吸支持),可用拮抗药时有效。 |
- 药物剂量与效应关系:药理效应与剂量在一定范围内成比例关系,这就是剂量-效应关系,简称量-效关系。
| 名词 | 定义 | 临床/研究意义 |
|---|---|---|
| 半数有效量(ED50) | 在给定的试验条件下,能引起50%实验动物出现既定阳性反应的药物剂量。 常用于描述药物效力 | 反映药物产生预期疗效所需剂量,值越小表示效力越高。 |
| 半数致死量(LD50) | 在给定的试验条件下,能导致50%实验动物死亡的药物剂量(median lethal dose)。 常用于毒性评价 | 反映急性毒性,值越大表示急性毒性越低(相对更安全)。 |
| 治疗指数(TI) | 通常定义为 TI = LD50 / ED50,用于表示药物的安全性范围。 TI越大,安全窗越宽。 | 作为安全性参考指标:TI大者相对安全。但单独用TI评估药物安全性并不完全可靠,应结合剂量–反应曲线、慢性毒性、个体差异和临床监测等因素综合评价。 |
- 药物与受体
| 项目 | 概念 / 机制 | 关键点 | 代表例子 |
|---|---|---|---|
| 受体激动药 | 与受体有亲和力并能激活受体产生反应(具有内在活性)。 | 按内在活性分:完全激动药与部分激动药 完全激动药:高亲和力+高内在活性,能产生最大效能。 部分激动药:虽有亲和力,但内在活性低,单独给药有效但达不到最大效能;与完全激动药同用可表现为对其的部分拮抗(竞争占位且内在活性较低)。 | 完全激动药:吗啡(μ受体完全激动) 部分激动药:喷他佐辛(pentazocine,阿片受体部分激动) |
| 竞争性拮抗药 | 与激动药竞争同一结合位点,结合可逆,按可逆竞争占位机制阻断受体激活。 | 剂量-反应曲线平行右移,最大效能(Emax)不变;可通过增加激动药剂量克服。 临床上用于快速可逆阻断或诊断(视药物半衰期与亲和力)。 | 阿片类:纳洛酮(naloxone) β受体:普萘洛尔(propranolol) 苯二氮卓:氟马西尼(flumazenil) |
| 非竞争性拮抗药 | 通过不可逆结合(共价)或别构(allosteric)阻断,或阻断离子通道功能,使受体不可被激活或能力下降。 | 不仅使剂量-反应曲线右移,且降低最大效能(Emax下降);增加激动药剂量通常无法完全克服。 机制包括不可逆结合(酶/受体永久失活)或通道阻断。 | 不可逆α拮抗:苯氧乙胺(phenoxybenzamine,不可逆α阻断) NMDA通道非竞争阻断:氯胺酮/PCP(ketamine/PCP) |
胆碱受体激动药、抗胆碱酯酶药与胆碱酯酶复活药
考点散乱。
考题
- 【例1】毛果芸香碱(匹鲁卡品)对眼的作用表现为:A. 降低眼内压、扩瞳、调节痉挛 B. 降低眼内压、缩瞳、调节痉挛 C. 降低眼内压、缩瞳、调节痉挛 D. 升高眼内压、扩瞳、调节痉挛 E. 升高眼内压、缩瞳、调节痉挛
- ✨【例2】临床上,新斯的明禁用于:A. 麻痹性肠梗阻 B. 机械性肠梗阻 C. 手术后尿潴留 D. 重症肌无力 E. 筒箭毒碱过量中毒
- 【例3】有机磷酸酯类急性中毒表现为:A. 腺体分泌减少、胃肠平滑肌兴奋 B. 膀胱逼尿肌松弛、呼吸肌麻痹 C. 支气管平滑肌松弛、唾液腺分泌增加 D. 神经节兴奋、心血管作用复杂 E. 脑内乙酰胆碱水平下降、瞳孔扩大
- 【例4】胆碱酯酶复活药不具备的药理作用是:A. 恢复已经老化的胆碱酯酶活性 B. 解除烟碱样症状 C. 恢复被抑制的胆碱酯酶活性 D. 与阿托品合用可发挥协同作用 E. 增高全血胆碱酯酶活性(内科学试题)
基础
胆碱受体激动药
- 毛果芸香碱的药理作用
| 药物 | 眼部主要作用 | 眼内压(IOP)与机制 | 调节(Accommodation) | 全身/局部分泌影响 |
|---|---|---|---|---|
| 毛果芸香碱 (Pilocarpine) Muscarinic agonist (M) | 缩瞳(miosis):激动瞳孔括约肌(主为M3),瞳孔缩小。 | 降低IOP:瞳孔缩小+睫状肌收缩→虹膜向中心牵拉、房角开大并通过小梁网/巩膜静脉窦(Schlemm管)促进房水外流。 | 睫状肌收缩→悬韧带松弛→晶状体变凸,适近视力↑,远视力↓(近视清楚,远视模糊)(调节痉挛)。 | 增加分泌:唾液、泪液、汗液、胃肠腺、胰腺及呼吸道粘液分泌增加。 |
| 阿托品 (Atropine) | 散瞳(mydriasis):拮抗瞳孔括约肌(M受体),瞳孔扩大;并阻断睫状肌M受体导致睫状肌松弛。 | 可升高IOP:瞳孔扩大时虹膜退向周边(尤其窄角眼),前房角变窄或发生瞳孔阻滞,阻碍房水回流至巩膜静脉窦,诱发或加重急性闭角性青光眼。 | 睫状肌麻痹→悬韧带紧张→晶状体变扁,适于远视力↑,近视力↓(远视清楚,近视模糊)(调节麻痹)。 | 减少分泌:唾液、泪液、汗液、胃肠腺、胰腺及呼吸道粘液分泌减少;可有口干、便秘、发热、心悸等抗胆碱症状。 |
- 毛果芸香碱的临床应用
| 适应症 | 用法与作用机制 | 要点 / 不良反应与注意事项 |
|---|---|---|
| 青光眼 | 低浓度滴眼液用于闭角性青光眼(环状或充血性青光眼),对早期开放性青光眼亦有一定疗效。 作用:缩瞳、扩大前房角间隙、降低眼内压 | 注意局部刺激、睫状体痉挛可致近视加重;不宜与强副交感药/眼内炎等合并使用前需评估;对有硫酸盐过敏或角膜疾病者慎用。 |
| 虹膜炎 | 与扩瞳药交替使用以防止虹膜与晶状体粘连(粘连预防)。 机制:通过调节瞳孔位置减少黏连风险 | 需监测眼压;炎症活动期使用时注意并发症风险;与其他眼用药物相互作用需评估。 |
| 其他系统性应用 | 口服可增加唾液分泌,用于放疗后口腔干燥症状缓解;亦可用于对抗阿托品(抗胆碱药)过量的解救治疗(作为胆碱能激动剂)。 | 常见副作用:出汗、流涎、胃肠蠕动增加、腹泻、心率减慢、支气管痉挛。哮喘、活动性消化性溃疡、严重心脏病或低血压患者禁忌或慎用。系统给药时注意剂量与监测生命体征。 |
易逆性抗胆碱酯酶药
拟胆碱作用
新斯的明
- 药理作用
| 作用分类 | 主要效应 | 机制 / 临床相关 |
|---|---|---|
| ① M 型(副交感/平滑肌、腺体) | 眼:缩瞳、降低眼内压 腺体分泌:增加汗、唾液、泪、胃肠腺、胰腺分泌 胃肠道:促进胃酸分泌;促胃、小肠、大肠蠕动,促排空 平滑肌:收缩支气管、输尿管等 心血管:心率减慢、心输出量下降;大剂量可致血压下降 | 由M受体激动或乙酰胆碱作用增强 → 副交感样反应。临床上与M受体介导的分泌和蠕动增强相关;用于缩瞳或促进肠胃动力时注意支气管/心血管副作用。 |
| ② N 型(神经肌接头/骨骼肌) | 对骨骼肌有双相效应:治疗剂量(例如新斯的明)→ 骨骼肌收缩力增强;过量→ 肌纤维震颤、肌束震颤,继而肌张力下降、肌无力或瘫痪 | 新斯的明为抗胆碱酯酶药,通过抑制AChE使神经肌接头处ACh增加,短期增强肌力(重症肌无力治疗),但过量ACh导致去极停滞或疲劳性阻断,出现毒性肌无力(胆碱能危象)。临床需区分剂量反应与监测呼吸肌功能。 |
- 临床应用
| 适应证 | 作用机制(要点) | 常见临床用途 & 给药要点 | 关键注意事项 |
|---|---|---|---|
| 重症肌无力(MG) | 抑制胆碱酯酶→ 增加突触间隙ACh浓度,作用于神经肌肉接头的骨骼肌型烟碱受体(nicotinic, N),改善肌力与耐疲劳性。 | 控制症状:口服或静脉。常用药物:新斯的明(neostigmine)或吡啶斯的明(pyridostigmine)。按需调整剂量以改善吞咽、眼肌与四肢肌力。 | 监测呼吸肌力以防肌无力危象与胆碱能中毒。结合免疫调节治疗。对ACh受体阻断以外原因需鉴别诊断。 |
| 术后肠胀气与尿潴留 | 通过增强外周胆碱能传递刺激平滑肌和膀胱逼尿肌,促进肠道蠕动和排尿。 | 常在术后出现肠瘫或尿潴留时给予静脉新斯的明并配合抗胆碱能(如阿托品)以减少副作用。注意评估机械性梗阻风险。 | 禁用于机械性肠梗阻或膀胱出口梗阻。可能引起腹痛、腹泻、支气管分泌增加、心率减慢(需并用阿托品或异丙阿托品按需拮抗心脏副作用)。 |
| 阵发性室上性心动过速(PSVT) | 短效提高迷走神经-心脏旁路胆碱能影响,可在特定情形下改变房室结传导。 | 临床上较少首选;迷走刺激或腺苷仍为首选。仅在特殊情况下由有经验人员评估后使用,需监测心电图。 | 对心脏传导病变慎用;可能诱发疝性心动过速、心动过缓或恶化心律失常。 |
| 非去极化型肌松药(如筒箭毒碱)过量逆转 | 抑制胆碱酯酶→ 增加ACh与骨骼肌型烟碱受体竞争性结合,从而逆转非去极化阻断恢复肌力。 | 术中/术后常用静脉新斯的明配合抗胆碱能(如阿托品或东莨菪碱)以防副作用;剂量按阻断程度调整并以肌力/呼吸指标为终点。 | 对去极化性肌松药(如琥珀胆碱)无效且可能加重。注意并用抗胆碱剂预防支气管/心脏副作用,必要时监测神经肌接头功能(TOF)。 |
难逆性抗胆碱酯酶药
有机磷酸酯类
有机磷酸酯类进入人体后,与乙 胆碱酯酶(AChE)牢固结合,形成难以水解的磷酰化 AChE,使 AChE 失去水解乙酰胆碱的能力,造成体内大量乙酰胆碱堆积而引起临床症状。
胆碱酯酶复活药
碘解磷定(PAM)
- 药理作用
| 作用类别 | 过程 | 关键要点 |
|---|---|---|
| 恢复胆碱酯酶活性(再活化) | 氧化咪唑类/阿托品配合的解磷定(如碘解磷定,oxime类)与被磷酰化的胆碱酯酶结合,形成中间复合物;随后断裂转移磷酸基团,使胆碱酯酶从被磷酰化状态恢复为可活化状态。 | 直接与被磷酰化的AChE结合并去除磷酸基,恢复酶活性 时窗重要:越早给药(未“老化”前)再活化成功率越高。 |
| 直接解毒(结合游离有机磷) | 解磷定可与体内未与酶结合的游离有机磷发生结合,生成被磷酰化的解磷定(无毒或毒性显著降低的产物),随后通过肾脏排出,减少游离毒物继续抑制胆碱酯酶的机会。 | 与游离有机磷直接结合,形成可排泄的无毒或低毒产物 临床意义:降低体内自由毒物负荷,辅助减少进一步酶抑制。 |
- 临床作用
| 作用/项目 | 具体说明 |
|---|---|
| 解除N样(烟碱样)症状 | 碘解磷定可显著减轻N样症状,对骨骼肌痉挛抑制最为明显,能快速抑制肌束颤动和肌强直,从而改善呼吸肌功能。 |
| 改善中枢神经系统症状 | 碘解磷定对中枢神经系统的中毒表现有一定改善作用,可减轻意识障碍、抽搐等症状,但效果有限且个体差异存在。 |
| 对M样(毒蕈样)症状的影响 | 对M样症状作用较小,故 有机磷农药中毒时应与阿托品合并使用,阿托品主要逆转M样(副交感)表现,二者联用可更全面控制症状。 |
M 胆碱受体阻断药、肾上腺素受体激动药与阻断药
考点散乱。
考题
- 【例1】具有缓解胃肠痉挛作用的自主神经递质受体阻断剂是 A. 阿替洛尔 B. 阿托品 C. 酚妥拉明 D. 育亨宾 E. 筒箭毒碱
- 【例2】术前准备时,常用于减少呼吸道腺体和唾液腺分泌的药物是 A. 山莨菪碱 B. 阿托品 C. 毛果芸香碱 D. 新斯的明 E. 去甲肾上腺素
- 【例3】能直接拮抗心脏迷走神经兴奋效应的药物是 A. 左旋多巴 B. 阿托品 C. 毛果芸香碱 D. 去甲肾上腺素 E. 酚妥拉明
- 【例4】能拮抗交感缩血管神经效应的药物是 A. 左旋多巴 B. 阿托品 C. 毛果芸香碱 D. 去甲肾上腺素 E. 酚妥拉明
- 【例5】能强烈收缩血管的药物是 A. 左旋多巴 B. 阿托品 C. 毛果芸香碱 D. 去甲肾上腺素 E. 酚妥拉明
- 【例6】临床上常作为升压药使用的药物是 A. 普萘洛尔 B. 去甲肾上腺素 C. 左旋多巴 D. 酚妥拉明 E. 肾上腺素
- 【例7】能减弱心肌收缩力并减慢心率的药物是 A. 普萘洛尔 B. 去甲肾上腺素 C. 左旋多巴 D. 酚妥拉明 E. 肾上腺素
- 【例8】由交感缩血管神经末梢释放的主要神经递质是 A. 普萘洛尔 B. 去甲肾上腺素 C. 左旋多巴 D. 酚妥拉明 E. 肾上腺素
- ✨【例9】使用过量氯丙嗪的精神病患者,如使用肾上腺素后,主要表现为 A. 升压 B. 降压 C. 血压不变 D. 心率减慢 E. 心率不变 解:氯丙嗪有明显的α受体拮抗作用,故给予肾上腺素时会出现“肾上腺素翻转”——α受体被阻断,β2介导的血管扩张占优势,导致血压下降(可伴有反射性心率加快,而非减慢)。
- 【例10】小剂量可以增加肾动脉血流量的药物是 A. 肾上腺素 B. 去甲肾上腺素 C. 异丙肾上腺素 D. 多巴胺 E. 氨茶碱
- 【例11】女,43岁。头晕伴心悸3天。查体:心率120次/分,血压150/93 mmHg。使用美托洛尔的药理作用是 A. 抑制迷走神经 B. 控制水钠重吸收 C. 抑制心肌收缩 D. 抑制肾素释放 E. 减弱心肌收缩力,减慢心率
- ✨【例12】妊娠患者最不宜选用的降压药 A. 利尿药 B. α受体阻断药 C. β受体阻断药 D. 血管紧张素转换酶抑制药 E. 二氢吡啶类钙通道阻滞药
- 【例13】哮喘患者最不宜选用的降压药为 A. 利尿药 B. α受体阻断药 C. β受体阻断药 D. 血管紧张素转换酶抑制药 E. 二氢吡啶类钙通道阻滞药
基础
M 胆碱受体阻断药(阿托品)
- 药理作用
| 系统 | 主要作用(机制) | 临床/生理后果(要点) |
|---|---|---|
| 眼 | 拮抗M胆碱受体:睫状肌松弛、瞳孔括约肌麻痹、虹膜括约肌功能丧失 | 散瞳(瞳孔扩大),调节麻痹(远视化,近用受限),晶状体变扁、屈光度降低;瞳孔扩大可导致虹膜后退、前房角变窄,房水回流受阻,眼内压升高(青光眼患者慎用)。 |
| 唾液与腺体分泌 | 阻断M受体:唾液腺、汗腺、呼吸道腺体分泌减少 | 口干、减少呼吸道分泌;可使体温调节受影响(尤其高温环境时)。 |
| 胃肠道 | 抑制胃肠平滑肌及腺体的M受体:降低平滑肌痉挛与蠕动,减少某些腺体分泌;对胃酸的直接抑制有限(不影响胃肠激素或非胆碱能递质引起的分泌) | 缓解胃肠痉挛、减轻绞痛;可导致便秘、胃排空延迟。对胃酸分泌抑制有限,不能完全替代其他抗分泌疗法。 |
| 胆道与子宫平滑肌 | 对胆道、子宫平滑肌的M受体作用较弱 | 临床效应有限,常不作为主要适应证。 |
| 泌尿系统 | 松弛膀胱逼尿肌、舒张输尿管/尿道平滑肌(通过M受体拮抗) | 可缓解膀胱痉挛和尿急,但也可引起尿潴留(尤其前列腺肥大患者注意)。 |
| 心血管 | 阻断迷走张力:心率增快(窦性心动过速);治疗量对血管/血压影响小 | 用于缓解迷走性心动过缓;注意心动过速和缺血风险。大剂量可能出现更强心血管反应。 |
| 中枢神经系统 | 低到中剂量可轻度兴奋延髓及高级中枢;较大剂量可能兴奋延髓、延链及大脑导致中枢症状 | 可能出现兴奋、谵妄、幻觉、意识改变;中枢作用随药物穿透血脑屏障能力与剂量而异(对穿透性强的制剂更显著)。 |
| 总体注意事项 | 作用基于M(胆碱能)受体拮抗,影响分泌、平滑肌、心率和瞳孔等 | 青光眼、前列腺增生、严重心血管疾病、重度高温/脱水、老年谵妄风险需慎用或禁用;监测心率、尿潴留与认知状态。 |
- 临床应用
| 适应/作用 | 要点与说明 |
|---|---|
| 解除平滑肌痉挛 | 对胃肠绞痛、膀胱刺激征疗效较好;对胆绞痛、肾绞痛疗效较差。 |
| 抑制腺体分泌 | 用于全身麻醉前给药以减少呼吸道和唾液腺分泌,防止分泌物堵塞呼吸道及吸入性肺炎;亦用于严重盗汗、重金属中毒、帕金森病导致的流涎症等。 |
| 虹膜睫状体炎 | 滴用0.5%–1% 阿托品溶液,松弛虹膜括约肌与睫状体,利于休息和炎症消退;可与缩瞳药交替使用以防粘连。 |
| 眼底检查与验光 | 滴眼使充分扩瞳,便于眼底检查;通过使睫状肌松弛并使晶状体固定,可准确测定屈光度(验光)。 |
| 抗缓慢型心律失常 | 用于迷走神经兴奋所致的窦房阻滞、房室阻滞等缓慢性心律失常的治疗。 |
| 抗休克 | 对暴发型流行性脑脊髓膜炎、中毒性菌痢、中毒性肺炎等导致的感染性休克,可用大剂量阿托品解除血管痉挛、扩张外周血管、改善微循环。 |
| 有机磷酸酯类中毒解救 | 主要用于解除其M样症状(腺体分泌过多、支气管痉挛、瞳孔缩小等)。 |
- 不良反应
| 分类 | 要点 | 备注 / 处理 |
|---|---|---|
| 常见不良反应 | 口干、视物模糊、心率加快、瞳孔扩大、皮肤潮红 | 轻—中度多为对症处理:补液、冷敷、监测生命体征;症状持续或加重时停药并评估。 |
| 中枢毒性 | 大剂量可致 运动失调、不安、激动、幻觉、谵妄、昏迷 | 及时识别为阿托品中毒;必要时入院监护、维持呼吸循环稳定、防压迫伤害。 |
| 特异性解救药 | 毒扁豆碱(physostigmine):缓慢静脉注射为首选解救药,可迅速逆转外周及中枢抗胆碱作用(包括谵妄与昏迷) | 给药前确认禁忌(如已发生严重支气管分泌、心律失常需慎用);准备救援设备并监测心电图与生命体征。 |
| 对症处理 | 苯二氮卓类(如地西泮)用于控制明显的中枢兴奋、惊厥或焦虑性激动 | 与毒扁豆碱合用时监测呼吸抑制与镇静深度;避免误用抗精神病药加重抗胆碱效应。 |
肾上腺素受体激动药
- 几种药物的机制对比
| 药物 | 类别 | α受体 | β1受体 | β2受体 |
|---|---|---|---|---|
| 去甲肾上腺素 (Norepinephrine) | α受体激动药 | +++ | ++ | +/- |
| 肾上腺素 (Epinephrine) | α、β受体激动药 | ++++ | +++ | +++ |
| 多巴胺 (Dopamine) | α、β受体激动药(低→高剂量效应不同) | + | ++ | +/- |
| 异丙肾上腺素 (Isoproterenol) | β受体激动药 | − | +++ | +++ |
去甲肾上腺素
- 药理作用
| 项目 | 要点 | 详细说明 |
|---|---|---|
| 受体与主要作用 | α受体(主要α1) β受体(主要β1,少数β2) | α1:外周血管收缩(以小动脉、小静脉为主);β1:兴奋心脏(心率↑、心肌收缩力↑、传导加速);β2:在部分血管可致舒张(如冠状动脉在心肌代谢↑时)。 |
| 血管分布与收缩程度 | 收缩最明显的部位 | 皮肤与黏膜血管收缩最明显,其次为肾、脑、肝、肠系膜、骨骼肌血管。小动脉与小静脉受影响最大。 |
| 对冠状动脉与心脏的影响 | 冠状动脉与心肌 | 尽管理论上收缩血管,但冠状动脉可因心肌兴奋和代谢产物(如局部代谢性扩张)而相对舒张。总体上心脏受β1兴奋显示收缩力和心率增加。 |
| 血压与心率的剂量相关效应 | 小剂量静脉滴注 | 血管收缩不显著,主要为心脏兴奋:收缩压↑、舒张压变化小、脉压增大。伴随血压上升可出现反射性心率减慢。 |
| 血压与心率的剂量相关效应 | 大剂量静脉滴注 | 明显外周血管收缩、外周阻力↑:收缩压与舒张压均升高、脉压变小;心排出量因射血阻力增加可不变或下降,且可能出现更明显的反射性心率变化。 |
| 代谢与其他系统影响 | 代谢与生殖系统 | 高剂量可致血糖升高(促糖原分解/糖异生);可增加孕妇子宫收缩频率;对中枢神经系统作用较弱(以周围作用为主)。 |
| 临床要点(简要) | 总结 | α介导:外周强烈收缩(皮肤、黏膜最显著)→ 血压↑;β1介导:心脏兴奋→ 收缩力↑、心率↑(但可被血压升高的反射性减慢部分抵消)。小剂量以心脏作用为主、大剂量以血管收缩与血压升高为主;并发血糖升高与对子宫的影响需注意。 |
- 临床应用
| 分类 | 要点 |
|---|---|
| 临床地位 | 已不再是休克一线治疗,临床应用范围明显缩小。 |
| 目前主要适应证 | 仅用于: • 早期神经源性休克(短期血压支持) • 嗜铬细胞瘤切除后低血压(术后短期支持) • 部分药物中毒导致的低血压(作为特殊情况的支持用药) |
| 给药途径及局部止血 | 本药在局部应用时可稀释后用于胃肠内操作(如内镜下注射/冲洗),能使食管和胃内血管收缩,产生局部止血作用。注意按科室/指南推荐的浓度与途径,避免系统性吸收导致的全身血流动力学变化。 |
- 不良反应
| 不良反应 | 机制 | 处理与预防 |
|---|---|---|
| 局部组织缺血坏死 | 静脉或皮下滴漏导致药物局部高浓度,引起血管强烈收缩、局部缺血坏死。 | 立即停止注射/更换部位;局部浸润注射普鲁卡因或α受体阻断剂(如酚妥拉明)以扩张血管;保持肢体温暖、抬高患肢并记录注射情况;严重者外科评估或皮瓣修复。 |
| 急性肾功能衰竭 | 滴注时间过长或剂量过大导致系统性血管收缩,肾血流减少,出现肾实质缺血/损伤。 | 监测尿量与肾功能,调整/停用可致血管收缩药物;避免长时高剂量滴注;必要时补液维持灌注压,早期肾科会诊与支持治疗(利尿、透析指征评估)。 |
肾上腺素
- 药理作用
| 系统/器官 | 主要受体 | 药理机制(要点) | 生理/临床效应 |
|---|---|---|---|
| 心脏 | β1(主要) | β1激动增加cAMP→ 正性肌力、正性频率、传导加速、自律性增强 | 心缩力↑、心率↑、传导速度↑;治疗量主要↑收缩压,舒张压不变或下降;大剂量两者均↑ |
| 血管 | α1(皮、黏膜、内脏 сосуды)、β2(冠脉、骨骼肌、肝) | α1→血管平滑肌收缩;β2→平滑肌舒张(局部与床位相关) | 皮肤/内脏血管显著收缩;冠状动脉、骨骼肌、肝血管舒张;脉压增大(治疗量);大剂量可使舒张压也升高 |
| 支气管/呼吸道 | β2(主要) | β2激动→平滑肌松弛;并可抑制肥大细胞脱颗粒 | 支气管舒张、减轻黏膜水肿、抗过敏(减少组胺释放) |
| 胃肠道 | α、β(平滑肌) | 总体抑制胃肠平滑肌收缩与运动(频率与幅度均减小) | 胃肠蠕动减弱、分泌活动下降 |
| 代谢(糖与脂) | β2(肝)、β(脂肪) | β2促进肝糖原分解;β刺激脂肪分解→血糖↑、游离脂肪酸↑ | 短期↑血糖与游离脂肪酸;临床上需注意糖代谢与高脂效应 |
| 中枢神经系统(CNS) | 多数儿茶酚胺(如肾上腺素) | 大多数肾上腺素类难以穿过血脑屏障;中枢作用与剂量相关 | 治疗量通常无中枢兴奋;大剂量可出现中枢兴奋症状(焦虑、顽固性兴奋等) |
| 药代/其他要点 | 剂量依赖性 | 相同药物在不同剂量可主导不同受体效应(低剂量常见β效应,较高剂量显α效应并出现更强的血压升高) | 临床使用时注意剂量、给药途径与靶床血流状态;警惕心肌需氧量增加与高血糖、心律失常风险 |
- 临床应用
| 作用/适应症 | 说明 / 关键点 | 给药途径与注意 |
|---|---|---|
| 心脏骤停(心脏复苏) | 肾上腺素为心肺复苏中首选药物,可在多种病因所致的心脏骤停中使用(如溺水、麻醉/手术突发事件、药物中毒、严重感染或高级传导阻滞导致的骤停)。电击/心室颤动时应以除颤为主,常与肾上腺素联合使用并配合高级心脏生命支持。 | 首选静脉给药(过去曾用心内注射,现多为静脉途径)。同时实施有效通气、胸外按压及纠正酸中毒等支持治疗。 |
| 过敏性疾病 | 一线抢救过敏性休克(过敏性休克时应迅速给予肾上腺素);用于控制急性支气管哮喘发作、缓解血管神经性水肿及某些血清病症状。 | 常见为皮下、肌注或静脉推注(视病情和剂量而定),注意监测心血管和神经系统副作用。 |
| 配伍(局部麻醉) | 局麻药中加入肾上腺素(常用浓度1:250000)可延缓吸收并延长麻醉作用,同时减少局部出血。 | 配伍时注意剂量与部位(末梢循环受限处慎用),避免全身过量吸收导致心动过速或高血压。 |
| 局部止血 | 可用于局部减少出血,但应慎用并少用,以免局部缺血或系统性吸收导致不良反应。 | 按需局部应用,监测局部组织灌注及全身反应。 |
| 治疗青光眼 | 肾上腺素可促进房水外流、降低眼内压,但在现代青光眼治疗中使用有限,应根据具体情况选择。 | 眼用制剂在眼科可短期使用,注意心血管副作用及对角膜的刺激。 |
- 不良反应
| 项目 | 要点 |
|---|---|
| 不良反应(概述) | 主要为:心悸、烦躁、头痛、血压升高等中枢及心血管症状。 |
| α受体过度激动 | 可导致血压骤升;严重者有发生脑出血危险,故老年人慎用,用药需谨慎并监测血压。 |
| β受体过度激动 | 可使心肌耗氧增加,诱发或加重心肌缺血和心律失常;故用量须严格控制,心脏病患者慎用并监测心率/心电图。 |
| 用药要点 | 掌握剂量、个体化调整;出现明显心悸、胸痛、血压显著升高或心律失常时应立即停药并处理。 |
多巴胺
- 药理作用:剂量依赖性
| 器官 | 药理机制 | 药理作用 |
|---|---|---|
| 心脏 | 高浓度(≥20 μg/kg)激动β1 受体 | 增强心肌收缩力、增加心排出量;导致收缩压和脉压升高 |
| 血管 | 高浓度(>20 μg/kg)激动α 受体;低浓度(≈10 μg/kg)激动D1 受体 | 高浓度:外周血管收缩、外周阻力增加,血压升高;低浓度:肾脏、肠系膜及冠状动脉舒张(血流增加) |
| 肾脏 | 低浓度激动D1 受体;大剂量可同时激动收缩性受体 | 舒张肾血管,增加肾血流和利尿;但大剂量时可使肾血管明显收缩(血流减少) |
- 临床应用
- 各型休克:如感染性休克、心源性休克、出血性休克等,应用前需补足血容量。
- ②急性肾衰竭:多巴胺与利尿剂联合用于急性肾衰竭,具有改善血流动力学的作用。
- 不良反应:一般较轻,偶见恶心、呕吐。
异丙肾上腺素
- 药理作用:血压效应与剂量相关
| 器官/系统 | 药理机制(受体) | 药理作用(要点) |
|---|---|---|
| 心脏 | 激动 β1 受体 | 正性作用:心肌收缩力↑、心率↑、传导速度↑ |
| 血管 | 激动 β2 受体 | 舒张骨骼肌血管、肾血管、肠系膜血管及冠状动脉 |
| 血压 | 效应与给药方式相关(静脉/肌注/吸入) | 静脉滴注可见收缩压↑且舒张压↓或略↓;静脉注射常见舒张压明显↓(与受体选择和剂量相关) |
| 支气管 | 激动平滑肌 β2 受体;对黏膜及肥大型细胞也有作用 | 支气管平滑肌舒张(缓解支气管痉挛);抑制肥大型细胞脱颗粒(减轻支气管炎症反应),但不能消除黏膜水肿 |
| 糖代谢 | 激动 α 和 β2 受体 | 促进糖原分解、升高血糖 |
| 脂代谢 | 激动 β3(及β2)受体 | 加速脂肪分解,增加游离脂肪酸释放 |
| 中枢神经系统(CNS) | 异丙肾上腺素类等过血脑屏障有限 | 中枢兴奋作用不明显(穿透血脑屏障差) |
- 临床应用
- ①支气管哮喘:用于控制支气管哮喘急性发作,舌下或喷雾给药,疗效快而强
- ②房室传导阻滞:舌下含化或静脉滴注给药,用于治疗二、三度房室传导阻滞
- ③心搏骤停:常为“心三联”的成分之一
- ④感染性休克:适用于中心静脉压高、心排血量低的感染性休克,但要注意补液
- 不良反应:心悸、头晕。
肾上腺素受体阻断药
α-肾上腺素受体阻断药 - 酚妥拉明
- 药理作用
| 部位/靶点 | 主要作用 | 机制 / 临床意义 |
|---|---|---|
| 血管 | 血压下降;外周阻力降低 | 通过阻断血管收缩受体并直接扩张小血管,导致周围阻力和动/静脉回流改变,临床上可出现低血压或直立性低血压。 |
| 心脏 | 心肌收缩力增强;心率加快;心输出量增加;可致心律失常 | 通过兴奋心脏(增加正性肌力与正性频率),可短期提高灌注,但增加氧耗并可能诱发或加重心律失常、心绞痛或心力衰竭。 |
| 胃肠(拟胆碱样) | 胃肠平滑肌兴奋,蠕动增加 | 具有拟胆碱能作用,可导致腹痛、腹泻、恶心、呕吐等副作用;对有活动性溃疡或肠梗阻者需谨慎。 |
| 胃酸(组胺样) | 胃酸分泌增加 | 通过组胺样作用刺激胃酸分泌,可能加重胃溃疡或反流相关症状。 |
- 临床应用
| 适应证 | 要点/机制及临床注意 |
|---|---|
| 周围血管痉挛性疾病 (如雷诺氏征、血栓闭塞性脉管炎、冻伤后遗症) | 扩张外周血管、缓解痉挛,用于改善肢体血流、减轻缺血症状;注意低血压与反射性心动过速风险。 |
| 去甲肾上腺素外渗 (静脉滴注外漏) | 局部皮下浸润酚妥拉明可局部扩张血管、逆转组织缺血;操作时注意剂量与局部组织反应,观察组织灌注恢复。 |
| 嗜铬细胞瘤 (高血压危象及术前准备) | 非选择性α受体拮抗,用于控制血压波动与减轻血管收缩,常与β阻滞(在α阻断充分后)联用;注意体位性低血压与反射性心动过速。 |
| 抗休克支持 (感染性、心源性、神经源性休克) | 扩血管、降低外周阻力可增加心排出量、改善肺循环并预防肺水肿,从而改善内脏灌注与微循环。使用时注意血流动力学监测,防止过度降压。 |
| 急性心肌梗死与难治性充血性心力衰竭 | 降低后负荷、增加心输出量以改善心功能和组织灌注;对血压、心率和缺血状况需密切监测,谨慎应用于有明显低血压者。 |
| 拟交感胺(如肾上腺素)过量引起的高血压 | 抗α作用逆转药物诱导的血管收缩,用于拟交感胺类药物过量所致的高血压;同时处理原发原因并监测循环动力学。 |
β-肾上腺素受体阻断药
β受体阻断药分非选择性和选择性两类。非选择性β受体阻断药可阻断β1+β2受体,如普蔡洛尔、吲哚洛尔等。选择性β受体阻断药可选择性阻断β1受体,如美托洛尔、阿替洛尔、艾司洛尔等。
- 药理作用
| 作用部位 | 药理机制 | 药理作用 |
|---|---|---|
| 心脏 | 阻断心脏的 β1 受体 | 负性变力与变时:心缩力↓、心率↓,心输出量和耗氧量减少 |
| 血管 | 阻断 β2 受体 | 肝、肾、骨骼肌及冠状动脉的血管收缩→局部血流量减少(选择性/非选择性差异明显) |
| 血压 | 作用与机体状态相关 | 对正常人血压影响小;对高血压患者可产生降压作用(外周阻力和心输出量共同作用) |
| 支气管 | 阻断支气管平滑肌的 β2 受体 | 支气管平滑肌收缩→可诱发或加重哮喘/支气管痉挛 |
| 糖代谢 | 对正常人影响有限;阻断 β 受体对糖代谢有关联 | 可延缓胰岛素后低血糖恢复(抑制糖异生和糖原分解的交感补偿反应) |
| 脂代谢 | 阻断β 受体 | 减少游离脂肪酸释放(影响脂肪动员) |
| 肾素-血管系统 | 阻断肾小球旁器的 β1 受体 | 抑制肾素释放→肾素-血管紧张素活性下降,利于降压 |
| 内在拟交感活性 (ISA) | 部分β受体阻断药同时具有 部分激动作用 | 对心率/血流的抑制较弱,临床上可减少严重心动过缓(如部分药物具有 ISA) |
| 膜稳定作用 | 降低细胞膜对离子的通透性(非β受体机制) | 某些β阻断药有局部麻醉样或奎尼丁样作用(可影响传导/兴奋性) |
- 不良反应
| 不良反应/风险 | 机制 | 临床要点(高危/处置) |
|---|---|---|
| 心血管抑制 | 阻断心脏β受体,降低心率、收缩力 | 可致心功能恶化、加重心力衰竭或诱发/加重房室传导阻滞。心衰、Ⅱ–Ⅲ度房室阻滞、严重窦性心动过缓者慎用或禁用;发生低输出表现应减量或停药并对症处理。 |
| 支气管痉挛/哮喘加重 | 尤其是非选择性β受体阻滞药,通过阻断β2受体导致支气管平滑肌收缩,增加气道阻力。 | 对有哮喘或慢性阻塞性肺病的患者应避免使用非选择性β阻滞剂,必要时选用高度β1选择性剂并慎用,出现呼吸困难立即停药并给予支气管扩张治疗。 |
| 反跳现象 | 长期用药后突然停药可因β受体上调引起症状回弹或加重(如心绞痛、心肌梗死、心率增快) | 应逐渐减量停药(循序减量数天至数周,视病情);既往冠心病或严重心血管病史者更需缓慢停药并监测。 |
| 其他中枢或皮肤不良反应 | 偶见眼-皮肤黏膜综合征、幻觉、失眠、抑郁等中枢神经或免疫相关不良反应。 | 出现明显精神症状或皮疹等异常,评估与药物相关性,必要时停药并对症处理。 |
局部麻醉药与镇静催眠药
丁卡因;苯二氮草类药理作用。
考题
- 【例1】丁卡因的作用或应用为 A. 可用于浸润麻醉 B. 脂溶性低 C. 穿透力低 D. 作用较普鲁卡因弱 E. 可用于表面麻醉
- 【例2】局部麻醉药普鲁卡因的特点是 A. 亲脂性强 B. 不易发生过敏反应 C. 毒性大 D. 容易成瘾 E. 对黏膜穿透力弱,不适合作表面麻醉(2021)
- 【例3】苯二氮卓类抗焦虑药物的主要作用为 A. 精神松弛 B. 肌肉松弛 C. 精神和肌肉都松弛 D. 阻断多巴胺受体 E. 阻断5-羟色胺受体
- 【例4】癫痫持续状态首选的治疗药物是 A. 苯妥英钠 B. 地西泮 C. 水合氯醛 D. 异戊巴比妥 E. 苯巴比妥钠
- 【例5】治疗脊髓损伤所引起的肌强直的药物是 A. 地西泮 B. 异丙嗪 C. 苯妥英钠 D. 氯丙嗪 E. 乙琥胺
- 【例6】治疗顽固性呃逆的药物是 A. 地西泮 B. 异丙嗪 C. 苯妥英钠 D. 氯丙嗪 E. 乙琥胺
基础
局麻药
| 项目 | 普鲁卡因 (Procaine) | 利多卡因 (Lidocaine) | 丁卡因/地卡因 (Tetracaine/Decacaine) |
|---|---|---|---|
| 别称 | 奴佛卡因 | 赛罗卡因 | 地卡因 |
| 所属类别 | 酯类局麻药 | 胺类/酰胺类局麻药 | 酯类局麻药 |
| 药理特点 | 脂溶性低,起效短,穿透力弱 对黏膜穿透力差 | 起效快、作用持久、穿透力强 临床最常用的局麻药 | 麻醉效能强、穿透力强 毒性较大 |
| 临床应用 | 浸润麻醉、传导麻醉、腰麻、硬膜外麻醉、局部封闭 | 全能型局麻药:用于浸润麻醉、传导麻醉、硬膜外麻醉 | 表面麻醉、传导麻醉、腰麻、硬膜外麻醉 |
| 不良反应 | 中枢神经系统和心血管反应、过敏反应(酯类代谢产物易致过敏) | 神经毒性较小,但可见中枢/心血管抑制与少数神经损害 | 毒性反应明显(系统吸收或过量可致严重中毒) |
| 注意事项 | 使用前需做皮试;不用于表面麻醉 | 适用于多数局麻场景;普鲁卡因过敏者首选 | 毒性大;一般不用于浸润麻醉(慎用于系统吸收部位) |
镇静催眠药
苯二氮卓类
与脑内苯二氮卓受体结合,促进Y-氨基丁酸(GABA)与GABA 受体结合,使CI通道开放的频率增加,导致更多的CI内流,从而增加了GABA 能神经的抑制效应。
- 药理作用
| 功能/适应证 | 要点 |
|---|---|
| ① 抗焦虑 | 选择性抗焦虑作用↑,小剂量即可明显改善焦虑症状。对多种原因所致焦虑疗效显著,主要用于焦虑症。作用机制:通过作用于边缘系统的苯二氮卓受体(GABA_A相关)实现。 |
| ② 镇静·催眠 | 随剂量增加出现镇静与催眠。缩短入睡潜伏期,延长总睡眠时间,减少觉醒次数;主要延长NREM第2期,对REMS影响较小。停药可见反跳性REMS延长(较巴比妥类轻)。依赖性与戒断症状相对较轻。可缩短NREM第3/4期,减少与该期相关的夜惊或梦游。 |
| ③ 抗惊厥(辅助) | 临床用于辅助控制破伤风、子痫、高热惊厥(小儿)、药物中毒所致惊厥等。 |
| ④ 抗癫痫 | 静脉注射为癫痫持续状态首选药物(例如地西泮),用于急性控制癫痫持续状态。 |
| ⑤ 中枢性肌肉松弛 | 具有较强的肌肉松弛作用,可减轻动物的去大脑僵直及人类因脑损伤导致的肌强直/僵硬。 |
| ⑥ 其他不良反应 | 大剂量可致短暂性记忆缺失(顺行性遗忘)、抑制肺泡通气(呼吸抑制)、血压下降与心率减慢等。 |
- 不良反应
| 项目 | 临床表现 / 风险 | 处理 / 注意事项 |
|---|---|---|
| 常见后遗效应 | 头晕、乏力、嗜睡、记忆力下降 | 注意:影响日间功能与驾驶风险增加 |
| 剂量相关毒性 | 大剂量可致共济失调;一次性大剂量口服或静脉注射过快可出现呼吸抑制、呼吸频率减慢、血压下降、循环衰竭 | 急救:维持气道、呼吸和循环;评估用药史 |
| 特异性拮抗剂 | 可用苯二氮卓类拮抗剂✨氟马西尼(flumazenil)用于鉴别诊断与抢救 | 慎用于有长期依赖或混合药物过量者(导致癫痫风险) |
| 药物相互作用 | 与其他中枢抑制药(如酒精、阿片类、抗组胺药、镇静抗精神病药)合用可加重中枢抑制 | 联合用药时严格评估抑制叠加和呼吸抑制风险 |
| 长期使用风险 | 长期大剂量应用可产生依赖性和成瘾;停药可出现戒断症状 | 逐渐减量,必要时结合心理或成瘾治疗支持 |
| 妊娠与哺乳期 | 长期用药可致畸形,妊娠早期禁用;产前与哺乳期应谨慎 | 育龄女性评估风险-益处,必要时选择替代方案 |
巴比妥类
- 药理作用
| 作用 | 药物/示例 | 要点 |
|---|---|---|
| 镇静‑催眠 | 小剂量:巴比妥类 | 小剂量:镇静,缓解焦虑与烦躁 中等剂量:催眠,缩短入睡潜伏、减少觉醒、延长总睡眠时间 |
| 抗惊厥 | 苯巴比妥(phenobarbital) | 具有强抗惊厥和抗癫痫作用 常用于广泛性大发作及癫痫持续状态的控制(急救/维持治疗中仍常用)。 |
| 静脉麻醉 | 硫喷妥钠(thiopental sodium) | 可作为静脉诱导麻醉药 起效快,作用短暂;注意呼吸抑制与心血管抑制风险。 |
抗癫痫药与抗惊厥药
癫痫治疗的首选药物
考题
- 【例1】苯妥英钠的不良反应不包括(2023) A. 牙龈损害 B. 共济失调 C. 肾损害 D. 过敏反应 E. 贫血
- 【例2】治疗癫痫小发作的首选药物是 A. 乙琥胺 B. 硫酸镁 C. 苯巴比妥 D. 扑米酮 E. 苯妥英钠
- 【例3】能治疗癫痫发作而无镇静催眠作用的药物是 A. 地西泮 B. 苯妥英钠 C. 苯巴比妥 D. 扑米酮 E. 以上都不是
- 【例4】对各型癫痫都有一定疗效的药物是 A. 乙琥胺 B. 苯妥英钠 C. 卡马西平 D. 丙戊酸钠 E. 苯巴比妥
- 【例5】三叉神经痛首选 A. 氟硝西泮 B. 苯妥英钠 C. 卡马西平 D. 氯丙嗪 E. 丙咪嗪
- 【例6】苯妥英钠不能用于治疗的病症是 A. 三叉神经痛 B. 舌咽神经痛 C. 癫痫局限性发作 D. 癫痫大发作 E. 癫痫小发作
基础
-
常考:
大英小二(乙)都有饼(丙)- 癫痫大发作首选苯妥英钠,小发作首选乙琥胺,大发作合并小发作首选丙戊酸钠。
- 癫痫局限性发作首选苯妥英钠,癫痫持续状态首选地西泮。
- 广请抗癫痫药物包括丙戊酸钠、卡马西平;丙戊酸钠对各型癫痫均有效。
- 三又神经痛、舌咽神经痛等中枢性疼病首选卡马西平,次选苯妥英钠。
-
硫酸镁主要用于缓解子痫、破伤风等的惊厥,也常用于高血压危象。
-
常用抗癫痫药的比较
| 抗癫痫药 | 别称 | 临床应用 |
|---|---|---|
| 苯妥英钠 | 大仑丁 | 首选:全面性大发作及局限性大发作/部分性发作;对小发作(失神发作)无效。用于三叉神经痛、舌咽神经痛及部分中枢痛综合征。亦为强心苷所致室性心律失常的首选药。 |
| 卡马西平 | (商品名:Tegretol) | 首选:大发作与单纯/复杂部分性发作之一;对神经性疼痛(如三叉神经痛)镇痛效果优于苯妥英钠。可用于难治性躁狂(锂盐无效时)及部分情感障碍。对尿崩症治疗无普遍指征(影像来源处有误时谨慎)。 |
| 苯巴比妥 | 鲁米那 | 用于癫痫大发作和癫痫持续状态(急救/维持控制),但不作为首选长期维持用药。对单纯/复杂部分性发作和精神运动性发作有效,对小发作无效。 |
| 普米酮 / 普米酮类 | (扑痫酮/Primidone) | 与苯巴比妥作用相近,临床上作为对其他药物控制不佳患者的替代选择。注意药代动力学与副作用谱。 |
| 乙琥胺 | (Ethosuximide) | 首选用于小发作(失神发作),副作用较少,专一性强,不适用于大发作首选。 |
| 丙戊酸钠 | 二丙基酰胺/Valproate | 广谱抗癫痫药,适用于多种癫痫类型(全面性与部分性均有效)。对大发作疗效优于苯妥英、苯巴比妥;对小发作和混合型(大发作合并小发作)是常用首选药。对部分难治性局灶性发作亦有作用。 |
中枢神经系统退行性疾病药与抗精神失常药
左旋多巴的作用机制及临床应用;氯丙嗪、碳酸锂、丙米嗪的作用机制及不良反应。
考题
- 【例1】左旋多巴的体内代谢特点是 A. 口服后主要在胃内吸收 B. 口服后大部分在肾内被吸收 C. 其在外周不能代谢为多巴胺 D. 其进入中枢后经多巴脱羧酶代谢失活 E. 口服后进入中枢的药物量很少
- 【例2】左旋多巴治疗帕金森病的药理机制主要是补充 A. 纹状体中左旋多巴的不足 B. 纹状体中多巴胺的不足 C. 黑质中左旋多巴的不足 D. 黑质中多巴胺的不足 E. 外周左旋多巴的不足
- 【例3】用左旋多巴或M受体阻断剂治疗震颤麻痹(帕金森病),不能缓解的症状是 A. 肌肉强直 B. 随意运动减少 C. 动作缓慢 D. 面部表情呆板 E. 静止性震颤
- 【例4】卡比多巴治疗帕金森病的机制是 A. 抑制中枢氨基酸脱羧酶的活性 B. 抑制外周氨基酸脱羧酶的活性 C. 抑制多巴胺的再摄取 D. 激动中枢多巴胺受体 E. 激动外周多巴胺受体
- 【例5】抗精神病药物的抗精神病作用的主要通路是 A. 锥体外系统 B. 结节-漏斗系统 C. 网状上行系统 D. 中脑-边缘系统 E. 黑质-纹状体系统
- 【例6】不属于氯丙嗪临床应用的选项是 A. 精神分裂症 B. 感染、中毒性精神病 C. 顽固性呃逆 D. 洋地黄引起的呕吐 E. 前庭刺激所致晕动症
- 【例7】多巴胺的临床应用是 A. 房室传导阻滞和甲状腺危象 B. 低温麻醉和人工冬眠 C. 异烟肼和链霉素治疗无效的结核病 D. 心源性休克和急性肾功能衰竭 E. 过敏性休克和支气管哮喘急性发作
- 【例8】氯丙嗪的临床应用是 A. 房室传导阻滞和甲状腺危象 B. 低温麻醉和人工冬眠 C. 异烟肼和链霉素治疗无效的结核病 D. 心源性休克和急性肾功能衰竭 E. 过敏性休克和支气管哮喘急性发作
- 【例9】碳酸锂中毒的早期症状为 A. 厌食、恶心、呕吐等胃肠道反应 B. 震颤、共济失调 C. 发热、定向障碍 D. 癫痫大发作 E. 下肢水肿、多尿
基础
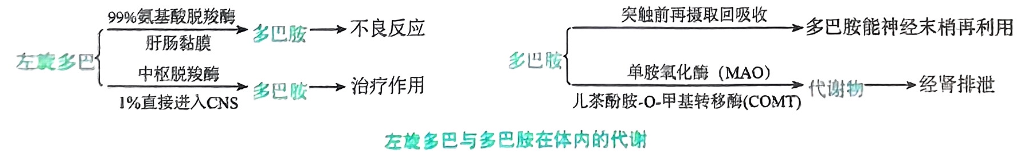
拟多巴胺药(左旋多巴)
-
机制
- 左旋多巴是由酪复酸形成儿茶酚胺的中间产物,为多巴胺的前体,需进入脑内转变为多巴胺才能发挥治疗作用。
- 同时合用L-芳香族氨基酸脱羧酶抑制药(如卡比多巴),可减少外周多巴胺的生成,使左旋多巴更多地进入脑内,转化为多巴胺而提高疗效
- 多巴胺不易通过血脑屏障,故不能用于治疗帕金森病
-
药理作用
| 项目 | 要点 |
|---|---|
| 适应证 | 治疗各型帕金森病(不限年龄、性别、病程);对由吩噻嗪类抗精神病药阻断多巴胺受体所致的帕金森样综合征无效。 |
| 起效与疗程 | 起效慢:用药后约2–3周出现体征改善,疗效逐步增强,通常在1–6个月达到最大效应。 |
| 疗效影响因素 | 疗效与黑质—纹状体病变程度相关:轻症及年轻患者效果较好;重症及老年患者效果较差。 |
| 对不同症状的效果 | 对肌肉强直与运动迟缓/困难改善明显;对震颤效果相对较差。 |
| 作用性质与局限 | 仅能缓解症状,不能阻止或逆转疾病进展。 |
- 不良反应
| 不良反应 | 发生率 | 临床表现 | 原因 | 处理 |
|---|---|---|---|---|
| 胃肠道反应 | 约80% | 恶心、呕吐、食欲不振 | 外周及中枢D2受体被刺激(多巴胺能药物或左旋多巴作用) | 给予胃动力/周围D2阻断药(如多潘立酮)或调整多巴胺药剂量 |
| 心血管反应 | 约30% | 直立性低血压、心律不齐 | 外周血管舒张/交感抑制或NA释放减少 | 监测血压/缓慢起立;必要时调整药物,应用β受体阻断或支持疗法 |
| 运动过度症(异动/舞蹈样动作) | 服药2年内高达≈90% | 异常不自主动作(手足、躯体、舌) | 长期多巴胺受体过度刺激导致运动通路异常兴奋 | 减量或分次调整多巴胺药;可试加胺类抗帕金森药(如金刚烷胺/阿曼他定)或使用抗多巴胺受体拮抗剂(谨慎) |
| 症状波动(on–off) | 3–5年后40%–80% | 症状突发加重/缓解交替(开—关反应) | 疾病进展导致左旋多巴储存/调节能力下降,药效不稳定 | 优化给药:分次小剂量、缓释制剂;可加多巴胺受体激动剂、MAO-B抑制剂、COMT抑制剂或持续肠外/肠内给药 |
| 精神症状 | 约10%–15% | 幻觉、妄想、情感抑郁或认知改变 | 多巴胺在皮质及边缘系统的过度或失衡作用 | 优先调整致病药物剂量;必要时使用非典型抗精神病药(如氯氮平)或低剂量喹etiapine,监测血象与副作用 |
左旋多巴增效药(卡比多巴)
- 卡比多巴又称洛得新,是外間复基酸脱羧酶抑制剂,单用基本无药理作用。
- 卡比多巴不易通过血脑屏障,与左旋多巴合用时,只能抑制外周氨基酸脱羧酶的活性,减少左旋多巴在外周转化为多巴胺的量,使进入脑内的左旋多巴增加
- 本药与左旋多巴组成的复方制剂称为心宁美,二者的混合比例为1:4或1:10
其它
- 溴隐亭系多巴胺受体激动药,与左旋多巴合用治疗帕金森病,能减少症状波动。
- 左旋多巴适用于各型帕金森病,但对吩噻嗪类抗精神病药所致的帕金森综合征无效。
- 苯海索是中枢性抗胆碱药,适用于轻症帕金森病,对抗精神病药所致的帕金森综合征有效。
抗精神失常药
抗精神分裂症药(氯丙嗪、氯氮平)、抗躁狂药(碳酸锂)、抗抑郁药(丙米嗪)和抗焦虑药(苯二氮卓类)。
经典抗精神病药(氯丙嗪)
整体上就是抑制性作用
-
冬眠灵,是吩噻嗪类药物的典型代表
-
药理作用
| 作用 | 药理机制 | 药理作用 |
|---|---|---|
| 抗精神病作用 | 拮抗脑内边缘系统及中枢多巴胺受体,主要为D2受体拮抗(并伴有不同程度的5‑HT2、α、H1受体作用,视药物而定)。 | 对中枢神经系统有较强抑制;对精神运动性兴奋、幻觉妄想等阳性症状疗效好;对情感淡漠、思维贫乏等阴性症状疗效较差(部分新药通过5‑HT2拮抗改善阴性症状)。 |
| 镇吐作用 | 小剂量通过阻断延髓第四脑室底部的化学感受区的D2受体发挥镇吐;大剂量可直接抑制呕吐中枢。 | 用于顽固性呕吐的控制;对前庭刺激或迷路性呕吐疗效差。 |
| 体温调节 | 强烈抑制下丘脑体温调节中枢(与多巴胺受体拮抗及α/5‑HT受体作用相关)。 | 既能降低发热性机体的体温,也可导致低体温;严重情况下可产生恶性综合征(需警惕)。 |
| 自主神经系统效应 | 阻断α受体、M胆碱受体及组胺H1受体(不同药物选择性不同)。 | 可导致血管扩张、血压下降(体位性低血压)、口干、便秘、视物模糊等抗胆碱及α阻断相关副反应。 |
| 内分泌影响 | 阻断下丘脑-垂体系统的D2受体,D2拮抗可促进垂体泌乳素释放;并影响促性腺激素、糖皮质激素和生长激素的分泌(与药物对下丘脑、垂体的综合作用相关)。 | 促使催乳素分泌↑(高泌乳素血症);抑制促性腺激素分泌,可见性功能改变;并可能影响糖代谢与生长激素/皮质醇轴,需长期监测。 |
- 临床应用
| 适应/用途 | 要点 |
|---|---|
| 精神分裂症(精神病性症状) | 对阳性症状(如攻击、亢进、妄想、幻觉)能显著缓解;对阴性症状(如冷漠、情感淡漠)效果不显著。 |
| 呕吐与顽固性呃逆 | 氯丙嗪有显著镇吐作用;对顽固性呃逆疗效明显;对晕动病(晕动症)无效。 |
| 人工冬眠(冬眠合剂)辅助治疗 | 冬眠合剂(由氯丙嗪、哌替啶和异丙嗪组成)用于短期诱导代谢下降与抑制应激反应,有利于机体度过严重缺氧或能量危机期,为进一步对因治疗争取时间。适应证:严重创伤、感染性休克、高热伴惊厥、中枢性高热、甲状腺危象等作为辅助治疗。 |
- 不良反应
| 不良反应 | 主要表现与处置要点 |
|---|---|
| 中枢抑制及抗胆碱/抗α受体效应 | 表现:嗜睡、淡漠、乏力;M受体拮抗:视物模糊、口干、少汗、便秘、眼内压升高;α受体拮抗:鼻塞、血压下降、直立性低血压、反射性心悸。处置:对症支持,必要时减量或停药,并纠正低血压;青光眼或严重便秘需专科处理。 |
| 锥体外系反应 | 帕金森综合征:肌张力增高、面容呆板、动作迟缓、肌震颤、流涎。可通过减少剂量或停药缓解;给予抗胆碱药(如苯海索/丙酰谷氨酰胆碱受体拮抗剂/二甲双胍类以外首选抗巴比妥类慎用)可改善症状。 |
| 静坐不能:坐立不安、反复徘徊;急性肌张力障碍:多见于用药后1–5天,表现为舌、面、颈、背部痉挛(伸舌、斜颈、呼吸受限、吞咽困难)。处置:立即小剂量抗胆碱药或肌肉松弛/静脉用药救治;必要时停药并观察。 | |
| 机制 | 由于氯丙嗪拮抗D2样受体(黑质—纹状体通路),使纹状体内多巴胺功能下降、乙酰胆碱相对增强,导致锥体外系反应。减量、停药或使用抗胆碱药可缓解。 |
| 精神与神经并发症 | 表现:意识障碍、萎靡、淡漠、兴奋、躁动等;应与原发精神病鉴别。发生者应立即停药并处理。少数出现惊厥/癫痫样放电,需抗癫痫处理并评估药物相关性。 |
| 过敏及罕见严重血液/肝脏损害 | 可见皮疹、接触性皮炎;少数可发生肝损害、黄疸、粒细胞减少、溶血性贫血、再生障碍性贫血等。出现血象或肝功能异常应停药并进一步检查。 |
| 心血管反应 | 直立性低血压、持续性低血压甚至休克,老年人、动脉硬化或高血压患者风险较高。监测血压,出现严重低血压给予升压支持并停药。 |
| 内分泌反应 | 长期用药可致高催乳素血症:乳房增大、泌乳、闭经、妊娠试验假阳性;可导致性功能障碍(性欲减退、勃起/生殖功能异常)及生长抑制(儿童)。处理:评估内分泌情况,考虑减量、换药或内分泌科评估。 |
| 急性中毒 | 表现:显著嗜睡、低血压、心肌损害(心动过速、心电图异常)等。需立即对症处理:维持气道通畅、循环支持、监测心电及电解质,必要时血液净化或重症监护。 |
非典型抗神经病药(氯氮平)
- 药理作用:氯氮平属于二苯并二氮卓类,为新型抗精神病药,几乎无锥体外系反应,可特异性拮抗中脑—边缘系统和中脑—皮层系统的D4受体,对黑质—纹状体系统的D2、D1受体几无作用。
- 不良反应:粒细胞减少,染色体畸变。
- 临床应用
| 项目 | 要点 | 说明 |
|---|---|---|
| 适应证 / 疗效 | 首选用于难治性精神分裂症 | 氯氮平(clozapine)对难治性病例效果明显,对既往对氯丙嗪等传统抗精神病药无效者仍可有效。可改善阳性与部分阴性症状。 |
| 起效速度 | 起效较快 | 通常在1周内可见临床改善(个体差异存在),起效较某些传统药物更快。 |
| 对慢性患者 | 对慢性及难治性患者有效 | 适用于长期慢性精神分裂症患者,尤其对其他药物无效者仍能获益。 |
| 迟发性运动障碍(TD) | 可用于既往典型抗精神病药致TD的治疗 | 在长期使用氯丙嗪等典型药物出现TD者,改用氯氮平可减轻运动症状,同时对原发精神病也有控制作用(证据支持但因个体差异需监测)。 |
| 局限与不足 | 对情感淡漠和形式逻辑思维障碍改善有限 | 氯氮平对情感淡漠(affective blunting)和思维组织障碍(如形式逻辑障碍)改善较差,需辅以康复、心理社会干预。 |
抗躁狂症药(碳酸锂)
- 药理作用
| 作用 | 机制 / 说明 |
|---|---|
| 抑制神经递质释放 | 在治疗浓度下,抑制神经元去极化并减少Ca2+依赖性突触前释放,特别降低去甲肾上腺素(NE)和多巴胺(DA)的释放;对5-羟色胺(5-HT)释放则不明显抑制,甚至可能相对保留或促进。 |
| 增加儿茶酚胺清除/灭活 | 促进突触间隙儿茶酚胺的摄取与灭活,降低突触间隙NE浓度,从而调节单胺能传递。 |
| 抑制细胞内第二信使途径 | 抑制腺苷酸环化酶(adenylate cyclase)及磷脂酶C(phospholipase C)介导的反应,改变cAMP、IP3/DAG等信号通路,影响细胞兴奋性与基因表达。 |
| 影响离子和代谢稳态 | 改变Na+, Ca2+, Mg2+在细胞内外的分布与可用性,并可影响细胞内信号转导和膜电位;同时可改变脑内葡萄糖代谢,影响能量供应与神经元功能。 |
- 临床应用
| 适应证 | 疗效与要点 |
|---|---|
| 躁狂症(急性/轻度) | 碳酸锂对躁狂症疗效显著,有效率约80%,对急性躁狂和轻度躁狂均适用,常作为一线情绪稳定剂。 |
| 抑郁症(双相抑郁/复发预防) | 主要用于双相障碍相关抑郁及抑郁复发预防。对抑郁的直接抗抑郁效果较躁狂弱,但长期使用可降低抑郁复发风险。 |
| 躁郁(双相)障碍 | 用于控制和预防躁狂与抑郁双相循环。长期规律用药可显著减少躁狂复发,且对抑郁复发也有预防作用,但对抑郁期效果不及对躁狂期。 |
- 不良反应
| 类别 | 临床表现 | 要点 |
|---|---|---|
| 轻度毒性 | 恶心、呕吐、腹痛、腹泻、细微震颤 | 出现上述症状时应评估用药史与剂量,考虑停药或调整剂量并监测血药浓度 |
| 重度毒性 | 精神紊乱、反射亢进、明显震颤、言语/发音困难、惊厥、昏迷、死亡 | 为危及生命情况,需立即停药、支持治疗并尽快住院;同时监测生命体征和血药浓度 |
| 血药浓度监测 | 血药浓度监测至关重要 | 当血药浓度>1.6 mmol/L时应立即停药并采取相应处理;若临床症状严重或浓度更高,考虑血液净化等措施 |
三环类抗抑郁药(丙米嗪)
- 药理作用
| 系统 | 药理机制 | 药理作用 |
|---|---|---|
| 中枢神经系统 | 阻断去甲肾上腺素、5‑HT 在神经末梢的再摄取,使突触间隙的递质浓度升高,促进突触传递。 | 患者连续服药后可出现精神振奋,约2–3周起效明显,情绪高涨、症状减轻。 |
| 自主神经系统 | 治疗量(如三环类或某些药物)可显著阻断 M 受体或胆碱能受体,导致胆碱能功能受抑。 | 可出现视物模糊、口干、便秘、尿潴留等副作用(即抗胆碱能效应)。 |
| 心血管系统 | 阻断单胺类神经递质的再摄取,导致心肌细胞去甲肾上腺素浓度增高;对心肌有奎尼丁样直接抑制效应(部分药物)。 | 可致血压下降或升高、心律失常、心动过速等;对心肌有直接抑制作用,需监测心电图与心功能。 |
- 临床应用
| 适应症 | 临床要点 | 备注(药理/使用提示) |
|---|---|---|
| 抑郁症 | 对内源性抑郁及更年期抑郁疗效较好;对反应性抑郁作用次之;对伴有精神病性症状的抑郁(即精神病性抑郁)疗效较差。另可用于强迫症的治疗,但目前首选为SSRIs。 | 常用三环类代表:丙咪嗪(imipramine)等;注意抗胆碱副作用、心电图改变、与其他抗抑郁/抗精神病药的相互作用及过量风险。 |
| 遗尿症(儿童) | 儿童夜间遗尿可试用丙咪嗪,对部分儿童有效,常作为短期或备选疗法。 | 用药前优先排除泌尿/内分泌病因;监测抗胆碱不良反应与心电图;避免长期连续用药。 |
| 焦虑与恐惧症 | 对伴有焦虑的抑郁症疗效显著;对特定恐惧症(如广场恐惧/特定恐惧)亦可有益。 | 三环类可有效缓解焦虑症状,但因耐受性与安全性问题,临床上常优先考虑SSRIs或SNRIs;合并用药需注意相互作用。 |
抗抑郁药(复西汀)
| 项目 | 要点 |
|---|---|
| 药理作用及机制 | 选择性5-羟色胺再摄取抑制剂(SSRI);抑制5-HT再摄取的效力约比对去甲肾上腺素再摄取强200倍。对肾上腺素受体、组胺受体、GABA受体、M型胆碱受体及多数5-HT受体几乎无亲和力。 |
| 临床应用 | 抑郁症;神经性贪食症(暴食型);强迫症。 |
| 不良反应 | 常见:恶心、呕吐、头痛、头晕、乏力、失眠;消化道和体重:厌食、体重下降;神经系统:震颤、可诱发或加重惊厥(罕见)。 |
镇痛药与解热镇痛抗炎药
往年很少考。 以后有机会再细看
考题
- 【例1】吗啡可引起 A. 瞳孔扩大 B. 呼吸抑制 C. 共济失调 D. 急性心力衰竭 E. 再生障碍性贫血
- 【例2】碳酸锂可引起 A. 瞳孔扩大 B. 呼吸抑制 C. 共济失调 D. 急性心力衰竭 E. 再生障碍性贫血
- ✨【例3】乙琥胺可引起 A. 瞳孔扩大 B. 呼吸抑制 C. 共济失调 D. 急性心力衰竭 E. 再生障碍性贫血
- 【例4】吗啡的适应证为 A. 哺乳期妇女止痛 B. 分娩止痛 C. 急性严重创伤疼痛 D. 颅脑外伤疼痛 E. 诊断未明急腹症疼痛
- 【例5】产妇临产前2~4小时内不宜使用的药物是 A. 哌替啶 B. 丙磺舒 C. 对乙酰氨基酚 D. 喷他佐辛 E. 布洛芬。 解:哌替啶(pethidine)和喷他佐辛均为阿片类或拟阿片镇痛药,能通过胎盘进入胎儿体内,产前2–4小时内使用可导致新生儿呼吸抑制、活动力差等不良反应,故不宜在临产近程使用。对乙酰氨基酚产时短程使用安全;丙磺舒与临产用药无直接禁忌;布洛芬等NSAIDs虽在妊娠后期长期或反复使用可引起动脉导管早闭,但在临产2–4小时内的主要禁忌并非本题所指。
- 【例6】吗啡和哌替啶的共同作用不包括 A. 体位性低血压 B. 止泻 C. 成瘾性 D. 镇痛 E. 抑制呼吸
- 【例7】非甾体抗炎药引起急性胃炎的主要机制是 A. 激活磷脂酶A B. 抑制弹性蛋白酶 C. 抑制前列腺素合成 D. 促进胃泌素合成 E. 抑制脂肪酶
- 【例8】既能治疗风湿性关节炎,又有抗血栓形成作用的药物是 A. 肝素 B. 布洛芬 C. 阿司匹林 D. 喷他佐辛 E. 哌替啶
钙通道阻滞药与抗心律失常药
钙通道阻滞药的使用原则;抗心律失常药的药理机制及应用。
考题
- 【例1】下列属于苯烷胺类选择性钙通道拮抗药的是 A. 硝苯地平 B. 维拉帕米 C. 普尼拉明 D. 哌克昔林 E. 氟桂利嗪
- 【例2】属于 Ic 类的抗心律失常药物是 A. 奎尼丁 B. 利多卡因 C. 普罗帕酮 D. 胺碘酮 E. 维拉帕米
- ✨【例3】对心房颤动无治疗作用的药物是 A. 强心苷 B. 奎尼丁 C. 利多卡因 D. 维拉帕米 E. 普萘洛尔
- 【例4】胺碘酮的药理作用是 A. 增加心肌耗氧量 B. 明显延长心肌不应期 C. 增加心肌自律性 D. 加快心肌传导 E. 收缩冠状动脉
- ✨【例5】便秘发生率最高的降压药是 A. 硝苯地平 B. 维拉帕米 C. 缬沙坦 D. 普萘洛尔 E. 卡托普利
- 【例6】具有抗心律失常、抗高血压及抗心绞痛作用的药物是 A. 可乐定 B. 普萘洛尔 C. 利多卡因 D. 硝酸甘油 E. 氢氯噻嗪
基础
钙通道阻滞药
-
类型
-
二氢吡啶类:硝苯地平、尼卡地平、尼群地平、氨氯地平、尼莫地平等。
-
苯并噻氮草类:地尔硫草、克仑硫草、二氯味利等
-
✨苯烷胺类:维拉帕米、加洛帕米、噻帕米等。
-
-
药理作用
| 靶点 / 效应 | 药理机制 | 药理作用 / 临床用途 |
|---|---|---|
| 负性肌力 | 钙通道阻滞使心肌细胞内 Ca2+ 减少 | 心肌收缩力下降,心肌耗氧量减少 |
| 负性频率 | 阻断房室结细胞 0 期、4 期去极化时由 Ca2+ 内流引起的活动 | 降低窦房/房室结自律性(减少心率) |
| 负性传导 | 通过抑制 Ca2+ 内流影响房室结传导 | 减慢房室传导速度(抗心律失常) |
| 普通血管 | 血管收缩所需的 Ca2+ 主要来自细胞外,钙通道阻滞可抑制该过程 | 扩张外周血管(主要对动脉作用明显),可舒张血管、降压 |
| 外周血管 | 松弛血管平滑肌,解除痉挛 | 用于治疗高血压、外周血管痉挛性疾病 |
| 脑血管 | 对脑血管舒张较敏感 | 扩张脑血管、增加脑血流量(如用于脑缺血相关情况) |
| 冠状动脉 | 舒张冠脉、减少冠状动脉阻力 | 增加冠脉血流,用于缓解心绞痛 |
| 平滑肌(气管、胃肠、泌尿、生殖道) | 松弛平滑肌(抑制 Ca2+ 依赖收缩) | 可舒张气管和平滑肌,较大剂量可影响胃肠、输尿管、子宫等 |
| 抗动脉粥样硬化 | Ca2+ 参与动脉粥样硬化病理过程,阻滞可干预该机制 | 具有抗动脉粥样硬化作用 |
| 红细胞稳定性 | 红细胞膜稳定性与 Ca2+ 密切相关,阻滞可抑制过量 Ca2+ 内流 | 抑制 Ca2+ 内流,减少 Ca2+ 超负载对红细胞的损伤 |
| 血小板 | 抑制血小板活化相关途径(与 TXA2 生成相关) | 抑制血小板聚集 |
| 肾功能 | 可扩张肾小血管,改善肾血流 | 保护/改善肾功能,降低肾动脉压力 |
抗心律失常药
- 分类

- 几种常考的药物的比较
| 药物 | 类别 | 作用机制 | 药理效应(要点) | 临床适应 |
|---|---|---|---|---|
| 利多卡因 (Lidocaine) | I b类 | 选择性阻断快速钠通道(处于去极或缺血组织更易结合) | 降低易动性,短缩或轻微降低动作电位时程(APD)、缩短有效不应期(ERP);对正常心肌影响小,对缺血心肌效用高 | 首选/常用于急性缺血相关的室性心律失常(室性早搏、室速、心肌梗死期室性危及生命性心律失常) |
| 普萘洛尔 (Propranolol) 及其他β阻滞剂 | II类 | 阻断β肾上腺受体(减弱交感激活) | 降低自律性,减慢房室结传导、延长AV结有效不应期,减少室率和心肌耗氧 | 房上性心律失常(室上速、房颤/房扑的率控)、预防心肌梗死后心律失常及减低猝死风险 |
| 胺碘酮 (Amiodarone) | III类(多靶点) | 主要阻断钾通道并兼阻钠、钙通道及α/β受体(多通路作用) | 显著延长APD和ERP,降低自律性,减慢传导;具有广谱抗心律失常作用,能扩张冠脉并降低心肌耗氧;长期用药有系统性毒性 | 广谱用于难治性室性/房性心律失常(复发性室速/室颤、持续性房颤控制、急性危及生命的心律失常);急救和长期维持均常用(注意肺、肝、甲状腺毒性) |
| 维拉帕米 (Verapamil) | IV类 | 阻断L型钙通道(主要在结间与慢反应组织) | 降低自律性,减慢AV结传导并延长AV结ERP,终止以AV结为通路的折返;对室肌作用弱 | 房上性心动过速、室上性折返性心律失常、对房颤房扑的率控有效(禁用于WPW合并房颤以避免房室旁路传导增强;慎用于严重心衰或低血压患者) |
利尿药与抗高血压药
各类降压药的机制及临床应用。
考题
- 【例1】主要作用于髓袢升支粗段、皮质部和髓质部的利尿药是 A. 螺内酯(安体舒通) B. 氢氯噻嗪 C. 甘露醇 D. 呋塞米(速尿) E. 氯噻嗪
- 【例2】心力衰竭合并肾衰竭患者首选的利尿药是 A. 阿米洛利 B. 氢氯噻嗪 C. 呋塞米 D. 螺内酯 E. 氯噻嗪
- 【例3】具有抗尿崩症作用的药物是 A. 氢氯噻嗪 B. 螺内酯(安体舒通) C. 甘露醇 D. 呋塞米(速尿) E. 50% 葡萄糖
- 【例4】某心源性水肿患者,用地高辛和氢氯噻嗪治疗,2 周后出现多源性室性期前收缩,其主要原因是 A. 低血钾 B. 低血钙 C. 低血钠 D. 高血镁 E. 低氯血症
- 【例5】女性患者,22 岁。在一次车祸中头部严重受伤,颅压升高。治疗方案中包括选用利尿药,不宜选用的药物是 A. 呋塞米 B. 甘露醇 C. 螺内酯 D. 布美他尼 E. 依他尼酸
- 【例6】可引起男子乳房女性化和妇女多毛症的药物是 A. 甘露醇 B. 螺内酯 C. 呋塞米 D. 糖皮质激素 E. 氨氯地平
- 【例7】静脉滴注甘露醇后引起利尿的性质是 A. 水利尿 B. 排钠性利尿 C. 保钾性利尿 D. 渗透性利尿
- 【例8】利尿药初期的降压机制为 A. 降低血管壁细胞内 Ca2+ 的含量 B. 降低血管壁细胞内 Na+ 的含量 C. 降低血管壁对缩血管物质的反应性 D. 排 Na+ 利尿,降低细胞外液和血容量 E. 诱导动脉壁产生扩张血管的物质
- 【例9】属于非二氢吡啶类钙通道阻滞药的是 A. 氨氯地平 B. 维拉帕米 C. 硝苯地平 D. 非洛地平 E. 吲达帕胺
- 【例10】高血压伴心绞痛及哮喘者,出现肾功能不全时,最适合的治疗药是 A. 卡托普利 B. 普萘洛尔 C. 硝苯地平 D. 氨氯噻嗪 E. 呋塞米
- 【例11】男,65 岁。高血压患者,使用依那普利控制血压不佳,改用氨氯噻嗪、螺内酯、硝苯地平、美托洛尔进行治疗,导致面色潮红、头痛。引起此并发症的药物是 A. 氨氯噻嗪 B. 螺内酯 C. 硝苯地平 D. 美托洛尔 E. 依那普利
- 【例12】血管紧张素转换酶抑制剂最适合于 A. 高血压伴双侧肾动脉狭窄 B. 高血压伴左心室肥厚 C. 高血压伴主动脉瓣狭窄 D. 高血压伴高钾血症 E. 妊娠期高血压
- 【例13】男,45 岁。慢性肾小球肾炎,高血压病史 3 年。规律服用血管紧张素转换酶抑制剂和螺内酯治疗,1 周前“上呼吸道感染”后出现尿量减少,近 2 天尿量约 100 ml/d。该患者最可能出现的电解质紊乱是 A. 血钾降低 B. 血镁降低 C. 血钙升高 D. 血钾升高 E. 血钠升高
- 【例14】高血压伴双侧肾动脉狭窄的患者降压不宜选用 A. 地尔硫卓 B. 硝苯地平 C. 氯噻嗪 D. 贝那普利 E. 美托洛尔
- ✨【例15】高血压伴痛风的患者降压不宜选用 A. 地尔硫卓 B. 硝苯地平 C. 氯噻嗪 D. 贝那普利 E. 美托洛尔
- ✨【例16】妊娠患者最不宜选用的降压药为 A. 利尿剂 B. α 受体拮抗剂 C. β 受体拮抗剂 D. 二氢吡啶类钙通道阻滞剂 E. 血管紧张素转换酶抑制剂
- 【例17】哮喘患者最不宜选用的降压药是 A. 利尿剂 B. α 受体拮抗剂 C. β 受体拮抗剂 D. 二氢吡啶类钙通道阻滞剂 E. 血管紧张素转换酶抑制剂
- ✨【例18】属于 AT1 受体阻断药的是 A. 尼群地平 B. 硝苯地平 C. 氯氮地平 D. 尼莫地平 E. 氯沙坦
- 【例19】在动物实验中,观察氯沙坦的药理作用主要通过测定 A. 肾素活性 B. ACE 活性 C. 尿量改变 D. 其抗 AT1 受体的活性 E. 血管平滑肌细胞内 Ca2+ 含量
基础
袢利尿药、噻嗪类药的利尿机制—抑制 NaCl 的重吸收(通过抑制 Na’-K’-2CI共转运子)。
乙酰唑胺的利尿机制——抑制 HCO;的重吸收(通过抑制碳酸酐酶活性)
螺内酯的利尿机制——竞争性抑制醛固酮受体(螺内酯为醛固酮的类似物)
作用部位——袢利尿药为髓袢升支粗段,噻嗪类为远曲小管,乙酰唑胺为近曲小管,螺内酯力远曲小管+集合管。
呋塞米、噻嗪类
| 项目 | 袢利尿药(Loop diuretics) | 噻嗪类利尿药(Thiazides) |
|---|---|---|
| 利尿效能 | 高效利尿,为最有效利尿药 | 中等效能利尿药 |
| 代表药 | 呋塞米(速尿)、依他尼酸、布美他尼 | 羟氯噻嗪(DHCT)、氯噻嗪、吲达帕胺、氯噻酮 |
| 作用部位 | 髓袢升支粗段(厚升支) | 远曲小管近端(远曲小管) |
| 作用机制 | 抑制髓袢粗段的Na⁺-K⁺-2Cl⁻共转运子(在Cl⁻结合位点),阻断NaCl重吸收 | 抑制远曲小管Na⁺-Cl⁻共转运子,减少NaCl重吸收 |
| K⁺排泄 | 远曲小管Na⁺负荷↑,促Na⁺-K⁺换位,使K⁺排泄增加(呈排钾型) | 同样因远曲小管Na⁺负荷↑,K⁺排泄增加(排钾型) |
| 电解质改变 | 尿中Na⁺、Cl⁻、K⁺、Mg²⁺、Ca²⁺均↑(促Ca排泄) | 尿中Na⁺、Cl⁻、K⁺↑,但Ca²⁺排出减少(保钙) |
| HCO3⁻排出 | 大量用药可抑制碳酸酐酶活性,HCO3⁻排出增加 | 轻度抑制碳酸酐酶,HCO3⁻排出略增 |
| 前列腺素(PG)相关 | 袢利尿药可促进局部PG合成,增强利尿和血流(非甾体药可抑制其效应) | 噻嗪类的某些作用依赖PG生成;非甾体可减弱其利尿作用 |
| 抗利尿作用(对ADH) | 无明显抗利尿作用 | 可明显减少尿崩症患者尿量并减轻口渴(治疗性作用) |
| 降压及血容量影响 | 可迅速降低充血、减轻静脉容量(用于急性肺水肿等),并降低外周容量 | 通过减少血容量与动力学改变降压;长期有轻度扩张外周血管作用(用于高血压) |
| 临床主要应用 | 急性肺水肿、脑水肿、重度水肿、急慢性肾衰伴容量超负荷、促药物或电解质排泄 | 轻–中度水肿、高血压、肾钙结石(预防Ca结石)、肾性尿崩症、内分泌性水潴留 |
| 常见水电紊乱 | 低钾、低钠、低氯、低镁、代谢性碱中毒、低血容量 | 低钾、低钠、低氯、低镁、代谢性碱中毒 |
| 高尿酸 | 可引起或加重高尿酸血症、痛风 | 可引起高尿酸血症、痛风者慎用 |
| 代谢影响 | 可致高血糖、高血脂(↑LDL、↑甘油三酯)、低HDL | 可致高血糖、高血脂 |
螺内酯
| 项目 | 要点 |
|---|---|
| 药理作用 | 醛固酮受体竞争性拮抗剂(螺内酯) 结构与醛固酮相似,阻断醛固酮效应:减少Na+重吸收和水潴留,促排钠排水、保留K+(即保钾性利尿)。起效缓慢:口服后约24小时起效,2–4天达最大效应;仅在体内有醛固酮存在时才显著发挥利尿作用(肾上腺切除动物无效)。 |
| 临床应用 | 利尿作用较弱、起效慢但持久。主要适应证: • 与醛固酮升高相关的顽固性水肿:肝硬化腹水、肾病综合征等(对低蛋白性或利尿难治性水肿尤为有效)。 • 充血性心力衰竭:通过利尿、钠潴留抑制及抑制心肌纤维化改善预后(亦用于慢性心衰长期治疗、降低病死率)。 • 原发性/继发性高醛固酮症可作为诊疗与治疗药物。剂量与联合用药依临床指征调整。 |
| 不良反应 | 常见:头痛、嗜睡、精神紊乱等中枢不良反应。 重要:• 高钾血症:保钾作用,肾功能不全、合并ACEI/ARB、K补充或高钾饮食者风险显著升高,需监测血钾与肾功能。 • 内分泌/性激素样副作用:可致男性乳房发育(gynecomastia)、性功能障碍;女性可有多毛(多见于螺内酯而非依替龙等选择性拮抗剂)。多为可逆,停药后常消失。 罕见/需警惕:过敏反应、严重电解质紊乱、肝功能异常等。 |
乙酰唑胺
| 项目 | 内容 |
|---|---|
| 药理作用 | 乙酰唑胺为碳酸酐酶抑制剂,主要在近曲小管抑制碳酸酐酶活性,减少HCO3−重吸收,导致尿碱化并有温和利尿作用。 核心:通过抑制碳酸酐酶降低HCO3−重吸收与房水生成 |
| 临床应用 | 总体:已较少单独作为常规利尿剂,用于若干特殊适应证。 |
| 青光眼:抑制眼内碳酸酐酶,减少房水生成,降低眼内压,对多类型青光眼有效(口服或局部制剂)。 | |
| 急性高山病:登山前约24小时开始口服,可减少脑脊液生成并使脑脊液/血液轻度酸化,减轻高山病相关头痛/水肿症状,用于预防与轻-中度治疗。 | |
| 碱化尿液:使尿液碱化,可促进尿酸、胱氨酸及某些弱酸性药物或毒物的溶解与排泄。 | |
| 纠正代谢性碱中毒:用于由过度利尿导致的代谢性碱中毒(短期协助纠正酸碱平衡)。 | |
| 其他适应证 | 可作为辅助治疗:癫痫(对部分难治性癫痫有作用)、伴低钾的周期性麻痹、严重高磷血症等(视具体病情与证据选择)。 |
| 不良反应 | 常见/重要:代谢性酸中毒(因丢失HCO3−)、低钾血症、疲乏、周围神经症状。其它:过敏反应、尿结石(碱化尿促进某些结石形成)、味觉改变、乏力。对青光眼或肾功能不全患者用药需权衡风险。 |
甘露醇
| 要点 | 机制 / 说明 | |
|---|---|---|
| 药理作用 — 脱水 | 静脉注射:迅速↑血浆渗透压,引起组织间液回流入血浆,产生组织脱水,降低颅内压与眼内压 口服:引起渗透性腹泻,用于胃肠道清除毒物 | 甘露醇为无法透过细胞膜的渗透性利剂;静脉给药在血管内保留渗透梯度,口服主要通过肠腔内高渗作用产生通便。 |
| 药理作用 — 利尿 | 增加尿量:增加循环量和肾小球滤过率,滤过后不被重吸收,减少髓袢升支与近曲小管水重吸收 | 静脉注射后短时扩容并直接在肾小管内维持高渗,抑制水的被动重吸收,从而产生渗透性利尿;需有一定肾灌注和滤过功能才能有效。 |
| 临床应用 | ① 脑水肿:首选药物,用于降低颅内压 ② 青光眼:急性发作或术前降低眼内压 ③ 急性肾衰竭:早期改善血流动力学变化,对伴低血压者更为有效 | 适应症应权衡体容积状态、电解质及肾功能;长期或大剂量可致脱水、血钠升高及肾功能负担,需监测血容、体重、电解质及尿量。 |
抗高血压药
| 药物类别 | 常用药物 | 药理作用 | 适应证 | 禁忌证 | 不良反应 |
|---|---|---|---|---|---|
| 利尿药 | 噻嗪类:氢氯噻嗪、氯噻酮 袢利尿:呋塞米 保钾利尿:螺内酯、阿米洛利 | 通过肾小管抑制不同部位的钠重吸收,减血容量、降低外周阻力,袢利尿作用强、起效快;保钾类拮抗醛固酮或抑制钠通道。 | 原发性高血压、伴有容量负荷或水肿的患者、心衰、低钾需保钾者(联合用药) | 严重低血容量、严重电解质紊乱、对某类药物过敏;保钾类对高血钾患者慎用 | 低钾、低钠、脱水、低血压、代谢性碱中毒(噻嗪)、高尿酸(痛风发作)、高血糖、听力损害(大剂量呋塞米)、高钾(螺内酯/阿米洛利) |
| 钙通道阻滞剂(CCB) | 二氢吡啶类:氨氯地平、硝苯地平、非洛地平 非二氢吡啶类:维拉帕米、地尔硫卓 | 阻断L型钙通道:血管平滑肌松弛,降低外周阻力;心脏型减慢心率和减少传导(非DHP)。二氢吡啶更强血管选择性。 | 高血压(尤其伴冠心病、外周血管疾病)、变异型心绞痛、部分老年高血压优先选择二氢吡啶;非DHP用于心率控制、房颤 | 严重低血压、代偿性心源性休克、重度主动脉瓣狭窄、已知药物过敏;非DHP在重度窦房/房室传导阻滞或充血性心衰时禁用/慎用 | 头痛、面部潮红、周围水肿(尤其二氢吡啶)、心悸、眩晕、便秘(维拉帕米)、心动过缓或房室传导阻滞(非DHP) |
| β受体阻滞剂 | 非选择性:普萘洛尔 β1选择性:美托洛尔、阿替洛尔 具有内在拟交感活性/α阻断:卡维地洛、拉贝他丁 | 阻断β1/β2受体:降低心率、心肌收缩力、心输出量;抑制肾素释放(部分),某些药有血管扩张附加作用。 | 合并冠心病、心绞痛、心肌梗死后二级预防、某些心律失常、部分甲亢症状控制;可用于高血压(常与其他药物联合) | 窦性心动过缓、2/3度房室阻滞(未植入起搏器)、急性心源性休克、未控制的失代偿性心衰、严重支气管哮喘(尤其非选择性β阻滞剂) | 乏力、冷汗、四肢末梢血流减少、心动过缓、恶心、勃起功能障碍、血糖掩盖(低血糖症状减少)、支气管痉挛(非选择性),停药需逐渐减量以避免反跳性心绞痛/高血压 |
| ACEI(血管紧张素转化酶抑制剂) | 卡托普利、依那普利、雷米普利、培哚普利 | 抑制ACE,减少Ang I→Ang II转化并↓降解缓激肽:降低血管收缩、降低醛固酮分泌,减小外周阻力和容量;抗重构作用 | 高血压、糖尿病合并蛋白尿/肾病、心衰、心肌梗死后心室重构预防 | 妊娠(绝对禁忌)、双侧肾动脉狭窄或单肾且依赖血管紧张素II维持肾灌注者慎用/禁用、既往ACEI引发的血管神经性水肿 | 干咳(与缓激肽相关)、高钾、低血压、肾功能恶化(起始或剂量增加时)、血管神经性水肿(少见但严重) |
| ARB(血管紧张素II受体阻断剂) | 缬沙坦、氯沙坦、厄贝沙坦、奥美沙坦 | 选择性拮抗AT1受体:阻断Ang II的收缩、醛固酮促分泌和重构效应,不增加缓激肽水平(较少干咳)。 | 高血压、心衰、蛋白尿/糖尿病肾病(ACEI不耐受时首选)、心肌梗死后部分指征 | 妊娠(绝对禁忌)、双侧肾动脉狭窄或对ARB过敏、严重低血压或血流动力学不稳定者慎用 | 高钾、低血压、肾功能恶化、少见血管神经性水肿(发生率低于ACEI)、头晕、乏力 |
治疗心衰的药物、抗动脉粥样硬化药与抗心绞痛药
各类药物的药理作用及临床应用。
考题
- 【例1】卡托普利抗心衰作用的机制是 A. 增加去甲肾上腺素分泌 B. 减少前列腺素合成 C. 拮抗钙离子的作用 D. 减少血管紧张素II的生成 E. 增加心肌耗氧量
- 【例2】治疗慢性心功能不全和逆转心肌肥厚并能降低病死率的药物是 A. 强心苷 B. 哌唑嗪 C. 硝酸甘油 D. 酚妥拉明 E. 卡托普利
- 【例3】美托洛尔降低心肌收缩力的机制是 A. β1受体激动剂 B. β2受体激动剂 C. β1受体阻断剂 D. β2受体阻断剂 E. M受体激动剂
- 【例4】急性左心衰竭合并房颤急性发作时,首选药物是 A. 胺碘酮 B. 强心苷 C. 奎尼丁 D. 维拉帕米 E. 利多卡因
- ✨【例5】强心苷对下列哪种原因所致的慢性心功能不全疗效较好? A. 甲状腺功能亢进 B. 维生素B1缺乏 C. 严重二尖瓣狭窄 D. 先天性心脏病 E. 缩窄性心包炎
- 【例6】强心苷治疗心房颤动的机制主要是 A. 缩短心房有效不应期 B. 减慢房室传导 C. 抑制窦房结功能 D. 直接抑制房性纤颤 E. 延长心房不应期
- 【例7】强心苷中毒最常见的心律失常类型是 A. 房性早搏 B. 室性早搏 C. 室性停搏 D. 房颤 E. 室颤
- 【例8】男,65岁。腹泻1周、心悸2天入院。既往有高血压、房颤病史。口服培哚普利、华法林、硝酸酯类、地高辛治疗。急诊心电图示频繁室性期前收缩、短阵室性心动过速。为明确患者病情改变的原因,应选择的检查是 A. 地高辛浓度测定 B. 凝血时间测定 C. D-二聚体测定 D. 血浆脑钠肽测定 E. 心肌坏死标志物测定
- ✨【例9】HMG-CoA还原酶抑制药的药理作用为 A. 抑制体内胆固醇生物合成 B. 阻断HMG-CoA转化为甲羟戊酸 C. 使肝脏LDL受体表达增强 D. 具有促进细胞分裂作用 E. 具有增强细胞免疫作用
- 【例10】男性,65岁。乏力、肌痛、酱油色尿1周。既往高脂血症病史10年,长期服用降脂药物。查体:体温36.5℃,脉搏89次/分,呼吸20次/分,血压140/85 mmHg。实验室检查:肌酸激酶1400 U/L。引起此不良反应的药物是 A. 考来烯胺 B. 非诺贝特 C. 阿托伐他汀 D. 烟酸 E. 依折麦布
- 【例11】女,70岁。冠心病、高血压、糖尿病患者。近1个月调整用药为阿司匹林、比索洛尔、辛伐他汀、二甲双胍。近3天双下肢无力及疼痛,双侧足背动脉搏动存在。实验室检查:血CK 2200 U/L,cTnI 0.01 ng/mL,血肌酐368 μmol/L。出现双下肢无力及疼痛的最可能原因是 A. 糖尿病足 B. 主动脉夹层 C. 间歇性跛行 D. 横纹肌溶解 E. 腰椎间盘突出症
- 【例12】硝酸甘油抗心绞痛的作用机制是 A. 增加心肌供氧量 B. 抑制心肌收缩力 C. 扩张外周血管 D. 减慢房室传导 E. 释放 NO
- ✨【例13】不属于硝酸甘油作用机制的是 A. 降低室壁张力 B. 降低心肌耗氧量 C. 扩张心外膜血管 D. 降低左室舒张末压 E. 降低交感神经活性
- 【例14】普萘洛尔与硝酸酯类合用治疗心绞痛的协同作用是 A. 增加心室容积 B. 降低心肌耗氧量 C. 加强心肌收缩力 D. 保护缺血心肌细胞 E. 松弛血管平滑肌
- 【例15】最可能加重变异型心绞痛的药物是 A. 抗血小板药物 B. 硝酸酯类药物 C. 钙通道阻滞药 D. 调脂药物 E. β受体阻断药
- 【例16】变异型心绞痛首选 A. 尼莫地平 B. 硝苯地平 C. 氨氯地平 D. 地尔硫卓 E. 尼群地平
- 【例17】男,57岁。心前区疼痛2年,多在安静状态发生,近日于睡眠中突发心前区疼痛。查心电图ST段抬高,冠脉造影见一过性狭窄。给予硝苯地平治疗的主要药理学依据是 A. 选择性增加心内膜下心肌供氧量 B. 选择性扩张冠状动脉,增加心肌供血 C. 显著减慢心率,降低心肌耗氧量 D. 显著抑制心肌收缩力,降低心肌耗氧量 E. 抑制或逆转心肌肥厚,降低心肌耗氧量
基础
血管紧张素转化酶抑制药(ACEI)
| 类别 | 要点 | 说明 |
|---|---|---|
| 代表药物 | ACEI | 卡托普利、依那普利、西拉普利、贝那普利、培哚普利、雷米普利、福辛普利 |
| 药理作用 | 降低外周血管阻力、减轻后负荷 | ACEI抑制血管紧张素转化酶,阻断 血管紧张素 I → 血管紧张素 II,减少血管收缩;同时抑制缓激肽降解,缓激肽增加可促进 NO和PGI2 生成,扩血管。 |
| 减少醛固酮生成、降前负荷 | 通过降低Ang II 减少 醛固酮 分泌,减轻水钠潴留,从而降低心脏前负荷。 | |
| 抑制心肌及血管重构 | Ang II 与醛固酮促进心肌细胞增生及间质纤维化,ACEI可防止并部分逆转心肌与血管重构。 | |
| 改善血流动力学 | 血管扩张、降低全身血管阻力,增加心搏输出量和运动耐力。 | |
| 降低交感神经活性 | 通过减少Ang II,抑制去甲肾上腺素释放和交感神经传递,具有抗交感作用。 | |
| 临床应用 | 心力衰竭一线药物 | 对各期心衰均有证据支持,可改善症状、减少住院及提高生存率;亦用于高血压、左室重构及某些肾病(慎用)。 |
| 不良反应/注意 | 常见与重要不良反应 | 首剂低血压、高血钾、干咳、血管性水肿、肾功能恶化;少见低血糖。用药前应评估血压、血钾、肾功能,利尿剂或脱水患者首剂前可先停/调整利尿剂并低起始剂量。妊娠禁用。 |
血管紧张素 II受体阻断药(ARB)
| 分类 | 要点 |
|---|---|
| 代表药物 | 氯沙坦、缬沙坦、厄贝沙坦、坎地沙坦、依普沙坦、替米沙坦、奥美沙坦(常简称为ARB) |
| 药理作用 | 直接选择性阻断血管紧张素II受体(AT1受体),阻断Ang II与受体结合→拮抗血管收缩、醛固酮释放及促增殖效应;能抑制/逆转心血管重构。 |
| 临床应用 | 用于高血压、各阶段心力衰竭(含射血分数降低者),常作为对ACEI不耐受(尤其因咳嗽)患者的首选替代药;亦用于糖尿病肾病的蛋白尿减少与肾保护(依指南与个体化评估)。 |
| 不良反应 | 总体不良反应少于ACEI,咳嗽发生率低;血管性水肿发生率也较低。其他:高钾血症、肾功能恶化(特别是肾动脉狭窄或低灌注时)、头晕、低血压。妊娠禁用(致畸/胎 nephropathy)。 |
β肾上腺素受体阻断药
| 项目 | 要点 | 详细说明 |
|---|---|---|
| 代表药物 | 非选择性/选择性 | 卡维地洛(carvedilol):β阻断+α1阻断、抗氧化;美托洛尔(metoprolol):β1选择性阻断。二者均为慢性心衰常用药。 |
| 药理作用与机制 | 抑制交感-肾素-血管紧张素系统(RAAS) | 阻断心肌与交感神经的β受体:降低儿茶酚胺毒性(减少过量儿茶酚胺引起的Ca2+大量内流、能量消耗和线粒体损伤),减少心肌细胞坏死与不良重构;抑制肾素释放,减少高浓度Ang II的心脏损害;长期可上调β受体数目并恢复信号转导与敏感性。卡维地洛另有α1阻断和抗氧化作用,抗交感更全面。 |
| 抗缺血与抗心律失常作用 | 减少心肌耗氧、稳定电生理 | 通过降低心率、收缩性和交感兴奋性,减轻心肌缺血并降低室性及致死性心律失常,是降低慢性心衰死亡率和猝死的重要机制。 |
| 临床应用 | 主要适应证 | 慢性收缩功能不全为主的心衰(尤其扩张型心肌病、缺血性慢性心衰)。长期规范使用可延缓症状恶化、改善心功能、降低猝死与心律失常发生率。 |
| 注意事项与禁忌 | 用药原则 | 起始剂量低、逐渐滴定至耐受或目标剂量;急性失代偿期需谨慎或短暂停用;对显著支气管痉挛、交感休克、严重窦房/房室传导阻滞应避免使用。 |
| 不良反应 | 常见/重点 | 窦性心动过缓、房室传导阻滞、低血压;可诱发或加重支气管痉挛(非选择性者更明显);精神抑郁、疲乏、性功能减退等。遇严重不耐受或明显过度减慢心率/低血压应减量或停药。 |
利尿剂
| 临床情景 | 推荐用药/方案 | 要点/备注 |
|---|---|---|
| 轻度充血性心力衰竭 | 单用噻嗪类利尿剂(如氢氯噻嗪、氯噻酮) | 适用于轻度液体潴留;监测血压、电解质与肾功能;对肾功能显著受损者疗效可能降低。 |
| 中-重度充血性心力衰竭 | 袢利尿剂(如呋塞米)或 噻嗪类 + 保钾利尿剂(如螺内酯、氨苯蝶啶)合用 | 袢利尿剂利尿强、起效快;合并保钾利尿剂可降低低钾风险,但需监测钾/肌酐及药物相互作用。 |
| 严重充血性心衰、急性肺水肿或全身严重水肿 | 静脉注射袢利尿剂(首选静脉呋塞米) | 噻嗪类口服常无效;静脉袢利尿剂起效迅速,需密切监测尿量、血压、电解质及肾功能;严重者联合血管扩张或正性肌力药视情形使用。 |
| 慢性心衰口服维持治疗 | 小剂量噻嗪或袢利尿剂,合并RAAS抑制剂/β受体阻滞剂等指南推荐药物 | 以症状和体重/液体状态为指导调整利尿剂剂量;避免过度利尿导致低血压或肾功能恶化。 |
| 主要不良反应 | 电解质紊乱、肾功能异常、高尿酸血症、耳毒性(主要为袢利尿剂)、代谢性异常 | 低钾血症为常见且临床重要问题(排钾利尿剂尤甚);监测指标:血钾、血钠、尿量、肌酐、尿酸;出现明显低钾或高钾时调整药物并补正电解质。 |
强心苷(地高辛)
强心苷是治疗心房扑动最常用的药物。
停用强心苷指征为视觉异常、心率 60次/分、低钾血症。
强心苷中毒最常见的不良反应是心律失常。
强心苷中毒最常见的心律失常是室性早搏。
强心苷中毒最常见的早期症状是胃肠道反应。
强心苷中毒的先兆是视觉异常(黄视、绿视、视物模糊)。
- 药理作用:强心、降传导
| 项目 | 要点(精简) | 机制 / 说明(精简) |
|---|---|---|
| 正性肌力 | 增强心肌收缩力、提高心排出量且相对不增加耗氧 | 抑制心肌细胞膜上Na⁺‑K⁺‑ATP酶→细胞内Na⁺↑→经Na⁺‑Ca²⁺交换使细胞内Ca²⁺↑,收缩力增强 |
| 负性频率 | 降低心率(对心衰伴快速室率者显著) | 强心剂增加每搏排出量→反射性兴奋迷走神经并提高心肌对迷走的敏感性,减慢心率与房室传导 |
| 自律性与传导性 | 低剂量降低窦房/房室结自律与房室传导;高浓度可增加自律性并致致律失常 | 低剂量以迷走反射为主;高浓度抑制Na⁺泵、细胞电生理改变(K⁺外流/细胞去极),最大舒张电位减小,易致心律失常 |
| 神经-胃肠毒性 | 中毒可致恶心、呕吐及中枢/延髓化学感受器兴奋 | 中毒刺激迷走及呕吐中枢;与电生理紊乱并存 |
| 心律失常风险 | 可诱发快速性或复杂型心律失常(中毒时常见) | 通过改变细胞内离子(Ca²⁺、Na⁺、K⁺)和交感/副交感平衡诱发室性或室上性异位节律 |
| 肾/内分泌效应 | 改善心输出量后肾灌注↑,肾素-血管紧张素活性↓,出现利尿 | 心功能改善→肾血流/滤过率增加→利尿;血中AT II及肾素水平下降 |
| 血管作用 | 可直接收缩血管平滑肌,外周阻力可升高 | 对外周血管有一定收缩作用,剂量及剂型相关 |
| 主要临床适应证(精要) | 慢性/急性心力衰竭(伴房颤或室率快者疗效最佳);控制室上性或部分室性心动过速;心房扑动时可转复或减慢室率 | 对因瓣膜病、冠心病等导致之心衰疗效较好;对肺心病、严重心肌损伤、广泛瓣膜狭窄或弥漫性心肌病疗效差;对多数房颤患者不能持续终止房颤,仅用于减慢室率或短期转复 |
- 不良反应
| 不良反应 | 临床表现或机制 | 防治措施 |
|---|---|---|
| 快速型心律失常 | 强心苷中毒时最常见且最早出现为室性早搏,可进展为室性心动过速或室颤。 | 静脉补钾(谨慎控制剂量,避免过量);合并传导阻滞者慎用或避免补钾;出现室性心动过速/室颤者首选利多卡因等抗心律失常药物并立即停用强心苷。 |
| 房室传导阻滞 | 强心苷可兴奋迷走神经、抑制窦房结或房室结,导致不同程度房室传导阻滞。 | 不宜盲目补钾;对有症状或高级房室阻滞者可用M受体拮抗药(阿托品);严重者需停药并考虑电复律或临时起搏。 |
| 窦性心动过缓 | 强心苷通过增强迷走张力可抑制窦房结,出现窦性心动过缓或停搏。 | 不宜补钾;可用阿托品改善症状;严重症状性心动过缓需停药并考虑临时起搏。 |
| 胃肠道反应 | 为常见早期症状,表现为厌食、恶心、呕吐、腹泻,可提示药物蓄积或中毒。 | 出现严重呕吐导致失钾者,应补钾或停药;轻中度者减量或停药并对症处理。 |
| 视觉异常 | 可见黄视、绿视、视物模糊等改变,常为强心苷中毒的提示性体征。 | 出现视觉异常应视为药物中毒征象,立即停药,必要时行血药浓度检测及支持治疗。 |
醛固酮受体阻断药(螺内酯)
| 项目 | 内容 |
|---|---|
| 代表药物 | 螺内酯(spironolactone)、依普利酮(eplerenone) |
| 药理作用 | 充血性心衰时血中醛固酮浓度显著升高。大量醛固酮除保钠排钾外,还具有明显的促生长作用:促进成纤维细胞增殖、刺激蛋白质与胶原合成,导致心房、心室及大血管的重构,加速心衰恶化。醛固酮受体拮抗剂通过阻断此通路,可减缓重构、改善血流动力学与临床症状并降低病死率。 |
| 临床证据 | 临床研究显示,在常规治疗基础上加用螺内酯或选择性受体阻断剂,可显著降低充血性心力衰竭患者病死率,并防止左室肥厚相关的心肌间质纤维化,改善预后。 |
| 临床应用 | ① 螺内酯:在充血性心衰中单药效应较弱,但与ACEI合用效果更佳,常用于减低病死率与抑制重构。 ② 依普利酮:为新型选择性醛固酮受体拮抗剂,高度选择性、耐受性好,能提高心衰患者生活质量并改善预后。 |
调血脂药与抗动脉粥样硬化药
HMG-CoA 还原酶抑制药(他汀类)
| 项目 | 要点 | 细节 / 机制 |
|---|---|---|
| 药理作用(降脂、血管保护、肾保护) | 降脂:竞争性抑制HMG‑CoA还原酶 | 他汀与HMG‑CoA结构相似,↓内源性胆固醇合成,显著↓LDL‑C,轻度↑HDL、↓TG |
| 血管及肾保护:多靶点非降脂效应 | 改善内皮功能(增强扩血管反应);抑制血管平滑肌细胞增殖/迁移、促凋亡;抗炎(↓CRP);抑制单核/巨噬细胞黏附与分泌;抗血栓(抑制血小板聚集、↑纤溶);抗氧化;减少泡沫细胞并稳定/缩小动脉粥样斑块;肾脏方面抑制系膜细胞增生、减轻炎症与免疫损伤,发挥肾保护作用 | |
| 临床应用 | 降脂为主;并用于心血管及肾脏保护 | 1. 家族性/非家族性Ⅱ型高脂血症及严重高胆固醇血症(可与其他调脂药合用)。 2. 2型糖尿病、肾病综合征相关高胆固醇血症(兼有肾保护)。 3. 预防心脑血管急性事件(↑斑块稳定性,↓卒中/心绞痛/AMI风险)。 4. 减少血管成形术后再狭窄、减轻移植后排异反应、辅助骨质疏松治疗等。 |
| 不良反应 | 常见轻度—少见重度 | 1. 暂时性:胃肠不适、肌痛、潮红、头痛(大剂量更常见)。 2. 酶学升高:可见无症状性转氨酶↑或CK↑,停药后通常恢复。 3. 严重:横纹肌溶解症(肌痛、肌无力、显著CK↑,罕见但需警惕)。 4. 动物资料:超大剂量可致犬白内障(临床相关性有限)。 |
贝特类
| 项目 | 要点 |
|---|---|
| 贝特类代表药物 | 吉非贝齐、非诺贝特、苯扎贝特 |
| 药理作用及机制 | 主要作用: 显著降低甘油三酯(TG)和极低密度脂蛋白胆固醇(VLDL‑C),并可降低总胆固醇(TC)和低密度脂蛋白胆固醇(LDL‑C),同时升高高密度脂蛋白胆固醇(HDL‑C)。 机制: 作为PPARα激动剂,上调脂蛋白脂酶(LPL)活性、促进VLDL清除、抑制肝脏甘油三酯合成和VLDL生成,并改善脂质代谢相关基因表达。作用强度与药物种类及剂型有关。 |
| 临床应用 | 主要用于以TG或VLDL升高为主的原发性高脂血症(如IIb、III、IV型高脂血症),尤其对高甘油三酯或混合型高脂血症疗效明显。注意与他药(如他汀)合用时的肌病风险并监测肝功能。 |
- 两者的比较
| 类别 | 代表药物 | 调脂机制 | 主要作用 | 其他作用 | 临床应用 | 不良反应 |
|---|---|---|---|---|---|---|
| 他汀类 (Statins) | 洛伐他汀、辛伐他汀、普伐他汀、阿托伐他汀 | 抑制 HMG-CoA 还原酶,降低肝内胆固醇合成;上调肝细胞 LDL 受体,增加 LDL 摄取与清除 | 降低血浆 LDL-C、总胆固醇,兼可降低 VLDL | 改善血管内皮功能,抑制平滑肌细胞迁移增殖,抗炎、稳定斑块,抑制血小板聚集,提升纤溶活性 | IIa、IIb、III 型高脂血症;2 型糖尿病或肾病综合征伴随的高胆固醇血症;预防心血管事件 | 胃肠道不适、肌痛/肌病、肝酶升高、偶见横纹肌溶解(风险随肌酶升高与药物相互作用增加) |
| 贝特类 (Fibrates) | 吉非贝齐、非诺贝特、苯扎贝特 | 激活 PPARα,促进脂蛋白脂肪酸代谢:减少肝内 TG 合成、降低 VLDL,促进 CM/VLDL 分解和 LDL 清除 | 显著降低 TG、VLDL-C,可降低总胆固醇和 LDL-C,升高 HDL-C | 具有 抗炎、抗凝 及改善微粒代谢作用 | 高甘油三酯血症(尤其原发性或混合型伴重度高 TG)、III 型(家族性混合)高脂血症;与糖尿病合并的高 TG | 消化道反应(恶心、食欲减退)、头痛、失眠,可与他汀合用时增加肌病风险;偶见肝酶升高 |
烟酸
| 项目 | 要点 |
|---|---|
| 药理作用 | 机制:为维生素B3(烟酸,niacin),在体内转化为NAD+/NADP+,参与氧化还原反应及能量代谢;对血脂有特异调节作用:抑制肝脏VLDL合成,减少LDL前体;显著提高HDL-C(通过抑制HDL受体介导的清除);还能降低甘油三酯和小而致病性LDL颗粒。 药效学特征:起效快(数小时内影响脂代谢),剂量相关性强;广谱代谢影响(肝、皮肤、糖代谢、尿酸)。 |
| 临床应用 | 适应证:主要用于高脂血症,尤其是合并低HDL-C的患者;用于: - 混合性高脂血症(特别是低HDL伴高TG) - 低HDL-C为主要问题的心血管风险管理(可作为他汀类或胆汁酸结合剂的联合用药) - 既往用于动脉粥样硬化性心血管病的二级预防(但近期大样本研究显示单独显著获益受限,需结合其他药物评估风险/收益)。 给药形式:即时释放(IR)、缓释(SR)和控释(CR/ER)制剂;联合他汀或胆固醇吸收抑制剂时常用。 |
| 不良反应 | 常见: - 面部潮红/瘙痒(flushing):机制为前列腺素介导,IR剂量相关,缓释或预先服用阿司匹林可减轻。 - 胃肠道不适(恶心、腹痛)。 严重/需监测: - 肝毒性(转氨酶增高至肝损伤,尤其为缓释高剂量时风险更高):需监测肝功能。 - 血糖升高:可使糖代谢异常加重、空腹血糖/糖化血红蛋白升高(糖尿病患者需谨慎)。 - 高尿酸/痛风诱发:可升高血尿酸。 - 肌病/横纹肌溶解(与他汀合用时肌病风险上升,应监测肌酶)。 - 罕见过敏反应、视网膜及皮疹报道。 |
| 用法与剂量(要点) | 起始常用剂量:500 mg/日,根据耐受性与疗效逐渐递增;常见维持剂量:1–2 g/日(分次给药);最大可达3 g/日但不推荐长期使用最大剂量。缓释或控释制剂可降低潮红但可能增加肝毒性风险。 合并他汀时:应从低剂量开始并监测肌酶、肝功能。 可用阿司匹林(75–325 mg)于给药前30–60分钟减轻潮红。 |
| 注意事项与禁忌 | 禁忌:活动性肝病或持续ALT/AST显著升高;严重肾功能不全(需个体评估)。 警示/监测:开始或剂量增加时每4–12周监测肝功能,随后按临床需要监测;糖尿病、痛风、肝病、活动性溃疡病或合并他汀者需加强监测;妊娠期禁用或谨慎(非必需维生素补充除外)。 药物相互作用:与他汀合用增加肌病风险;与降糖药、利尿剂等可能相互影响代谢参数。 |
| 临床实践建议(简明) | 将烟酸用于以低HDL为主的脂代谢异常或他汀不能充分纠正低HDL时的补充方案;开始低剂量、逐步递增并监测肝功能、血糖、尿酸与肌酶;潮红影响依患者可用阿司匹林预防或改用缓释制剂但权衡肝毒性风险。 在心血管终点方面,当前证据提示需与他汀等一线药物综合评估治疗获益与风险,单独应用并非首选。 |
胆固醇吸收抑制剂(依折麦布)
| 项目 | 详细说明 |
|---|---|
| 药理作用 | 依折麦布(ezetimibe)选择性抑制小肠刷状缘NPC1L1蛋白介导的胆固醇吸收,减少肠腔胆固醇进入肝脏,导致肝细胞LDL受体上调,增加血浆LDL‑C清除。对膳食与胆汁胆固醇均有效;作用靶点非HMG‑CoA还原酶,与他汀机制互补。 |
| 药代动力学 | 口服后在肝肠系统被葡萄糖醛酸化为活性代谢物(ezetimibe‑glucuronide);峰浓度约1–2小时;与食物影响小;主要经粪便排泄,少量经尿;活性代谢物半衰期约22小时,支持每日一次给药。 |
| 临床适应证 | 原发性高胆固醇血症(轻–中度;包括遗传性家族性高胆固醇的某些患者);与他汀合用用于未达LDL‑C目标者或耐受性差者;单用用于对他汀禁忌或不能耐受者;也可作为混合型血脂异常的补充治疗。 |
| 疗效与联合策略 | 单药可使LDL‑C约下降15%–20%(个体差异)。与他汀合用可在基线他汀治疗上额外获得约相对15%左右的LDL‑C进一步下降,常用于强化降脂。与胆汁酸结合树脂、PCSK9抑制剂等联合须个体化选择。 |
| 给药与剂量 | 推荐口服10 mg,每日一次,可与他汀同时或分开服用(给药时间不受限)。肾功能不全通常无需调整;严重肝功能损害慎用或禁用。 |
| 主要不良反应 | 常见:上呼吸道感染、腹痛、腹泻、肌痛(发生率接近安慰剂)。罕见:转氨酶升高、胆囊疾病(结石/炎症)、严重过敏反应。与他汀合用时肌病/横纹肌溶解的风险虽低但需关注,尤其在合并使用增加他汀暴露的药物时。 |
| 禁忌与警示 | 禁忌:对依折麦布或辅料过敏者禁用。 警示:既往严重肝病或持续性转氨酶升高者慎用;妊娠与哺乳期不推荐(数据不足);合用他汀时需监测肝功能与肌酶。 |
| 药物相互作用 | 总体相互作用较少。与胆汁酸结合树脂同时服用会降低依折麦布吸收,建议树脂服用后4–6小时再服依折麦布。与他汀联用无明确药代学上显著不良互作,但临床上需警惕肌病风险,注意与强效CYP3A4抑制剂或其他能增加他汀浓度的药物联合时的风险。 |
| 监测建议 | 开始前评估基线血脂、肝功能;如有肌痛史或高危者评估CK。用药后4–12周复查血脂评估疗效并调整方案;出现肌痛或无力测CK;若与他汀合用且转氨酶持续升高≥3×ULN,考虑停药或调整治疗。 |
| 特殊人群 | 肾功能不全无需常规剂量调整;中度肝功能受损者慎用,严重肝病禁用。妊娠与哺乳期不推荐。儿童在专科评估下可用于某些遗传性高胆固醇病例,并需随访。 |
| 临床应用要点 | 对无法达标的高危患者,可在最大耐受他汀基础上加用依折麦布以进一步降低LDL‑C;对他汀不耐受者可考虑依折麦布单药或与其他非他汀药物联合。评估心血管结局获益需结合患者基线风险及最新循证证据个体化判断。 |
| 临床证据与效果评估 | 多项RCT显示依折麦布能显著降低LDL‑C,且与他汀联合时提供附加降脂效果。大型结局研究与指南支持在特定人群中合并治疗可降低心血管事件风险,但具体获益受基线风险、随访时长和共用药物影响,应参照最新指南并个体化决策。 |
抗心绞痛药
硝酸酯类(硝酸甘油)
- 不影响交感神经活性
| 类别 | 要点 | 具体说明 |
|---|---|---|
| 药理作用 | 平滑肌松弛 | 主要对血管平滑肌:小剂量优先扩张静脉,减低回心血量和心肌耗氧;较大剂量兼扩张动脉,降低射血阻力和后负荷;降低左室充盈压、改善心内膜灌注与左室顺应性;可保护缺血心肌(促进内源性保护因子释放)。 |
| 作用机制 | NO供体 → cGMP ↑ | 硝酸甘油经谷胱甘肽转移酶在平滑肌细胞内释放一氧化氮(NO),激活鸟苷酸环化酶使cGMP↑,降低细胞内Ca2+释放与外流入,致血管平滑肌松弛。另促使PGI2、降钙素基因相关肽释放,抑制血小板聚集/黏附,参与冠状动脉与侧支循环扩张。 |
| 临床应用 | 心绞痛、AMI、心衰、肺血管病 | 1) 心绞痛:舌下制剂起效快,发作前预防可用。 2) 急性心肌梗死:常用静脉给药,降低耗氧、改善缺血灌注并抑制血小板;注意限制连续用量以免过度低血压。 3) 心力衰竭:降低前后负荷,改善血流动力学。 4) 急性呼吸衰竭/肺动脉高压:可舒张肺血管、降低肺血管阻力并改善通气-灌注。 |
| 不良反应与注意 | 低血压、头痛、潮红 | 常见为血管舒张相关:面部潮红、搏动性头痛、头晕、体位性低血压。需监测血压,避免与PDE5抑制剂合用(可致严重低血压);连续或反复用药可发生耐受,须间断或调整方案。 |
钙通道阻滞药
- 硝苯地平具有显著的扩张冠脉和外周小血管的作用,首选用于变异型心绞痛。
- 维拉帕米扩张冠脉的作用较弱,对变异型心绞病不宜单独使用;对稳定型心绞痛有效,但不是首选。
- 地尔硫卓对变异型、稳定型和不稳定型心绞痛均有效,但首选用于稳定型心绞痛。
| 项目 | 要点 |
|---|---|
| 抗心绞痛作用 | 机理(通过阻滞电压依赖性钙通道,减少Ca2+内流) 1. 降低心肌耗氧量:通过减弱心肌收缩力、减慢心率并降低外周血压,减轻心脏前后负荷,从而降低耗氧。 2. 舒张冠脉:尤其能解除冠脉痉挛(对变异型心绞痛作用显著)。 3. 保护缺血心肌:抑制缺血时的细胞内Ca2+超载,减轻钙毒性相关损伤。 4. 抑制血小板聚集:通过降低血小板内Ca2+浓度,抑制聚集(辅助抗栓作用)。 |
| 临床应用 | 主要适应证与注意 1. 变异型(Prinzmetal)心绞痛:首选或最佳适应证,因能解除冠脉痉挛。 2. 稳定型心绞痛与急性心肌梗死:可作为抗缺血治疗(与其他药物联合以优化血流动力学)。 3. 合并哮喘者:比β受体阻滞剂更合适(可舒张支气管平滑肌)。 4. 对心力衰竭诱发较少:同等剂量下对心肌抑制相对较弱,但在已有重度收缩功能不全时仍需谨慎使用(避免过度负荷下降或窦性心率过慢)。 |
作用于血液及造血器官的药物与组胺受体阻断药
考点乱
考题
- 【例1】具有体内、外抗凝血作用的药物是 A. 肝素 B. 阿司匹林 C. 香豆素类 D. 链激酶 E. 右旋糖酐
- 【例2】抗凝血酶的作用是 A. 封闭凝血因子活性中心 B. 抑制前列腺素合成 C. 抑制血小板聚集 D. 抑制TXA2形成 E. 增加纤维蛋白溶解
- 【例3】可减弱香豆素类药物抗凝血作用的药物是 A. 甲苯磺丁脲 B. 奎尼丁 C. 阿司匹林 D. 口服避孕药 E. 羟基保泰松
- 【例4】下列药物中,属于肝药酶抑制药的是 A. 利福平 B. 苯妥英钠 C. 苯巴比妥 D. 西咪替丁 E. 双香豆素
- 【例5】链激酶属于 A. 促凝血药 B. 纤维蛋白溶解药 C. 抗贫血药 D. 抗血小板药 E. 补血药
- 【例6】叶酸可以治疗 A. 地中海贫血 B. 缺铁性贫血 C. 巨幼细胞贫血 D. 溶血性贫血 E. 出血
- 【例7】女,25岁。因过敏性鼻炎服用抗组胺药后出现严重的嗜睡、困倦、口干。最可能服用的药物是 A. 西替利嗪 B. 非索非那定 C. 氯司咪唑 D. 氯雷他定 E. 氯苯那敏(2024)
- 【例8】雷贝拉唑的主要作用是 A. 中和胃酸 B. 促进胃排空 C. 抑制胃酸分泌 D. 黏膜保护作用 E. 阻断促胃液素受体
- 【例9】雷尼替丁的主要作用是 A. 中和胃酸 B. 促进胃排空 C. 抑制胃酸分泌 D. 黏膜保护作用 E. 阻断促胃液素受体
基础
肝素类抗凝血药(肝素)
| 项目 | 要点 | 机制 / 备注 |
|---|---|---|
| 药理作用 | 抗凝 调节血脂 抗炎 抑制血管内膜增生 抑制血小板聚集 | - 抗凝:通过增强抗凝血酶Ⅲ(ATIII)对凝血酶(IIa)及凝血因子Xa、IXa、XIa等的抑制,加速ATIII‑酶复合物形成。静脉注射后数分钟起效,APTT在10min内明显延长,作用持续约3–4h(对未分馏肝素)。 - 调脂:刺激血管内皮释放脂蛋白脂酶,水解乳糜微粒和VLDL。 - 抗炎/抗增生:抑制炎性介质与炎症细胞活性,抑制血管平滑肌细胞增殖。 - 抑制血小板聚集:部分为继发于凝血酶抑制,也有直接影响。 |
| 临床应用 | 主要适应证(要点) | - 防治静脉与动脉血栓栓塞:深静脉血栓(DVT)、肺栓塞(PE)。 - 弥散性血管内凝血(DIC):各种病因所致DIC的主要适应证之一(根据病情与出血风险权衡)。 - 急性冠脉/脑梗死的抗凝(在指征下)及术中/术后抗凝:心血管手术、外周血管手术、心导管操作、体外循环、血液透析等体外抗凝。 |
| 不良反应 | 出血 血小板减少(含肝素诱导性血小板减少症 HIT) 过敏反应 长期用药:骨质疏松 | - 出血为首要风险,需按APTT/临床评估调整剂量并备逆转措施(如硫酸二甲酰胺/脯氨酸?请按局部指南)。 - HIT:免疫介导,出现进行性血小板下降并伴血栓风险,应立即停用肝素并改用非肝素抗凝剂。 - 长期大剂量使用可引起骨质疏松与压缩性骨折。 |
香豆素类抗凝血药
| 项 | 内容 |
|---|---|
| 常用药物 | 双香豆素(dicoumarol)、华法林(warfarin)、醋硝香豆素(acenocoumarol)等 |
| 药理作用与机制 | 抑制维生素K循环:阻断肝内维生素K环氧化物还原为氢醌(不能再生利用),从而降低维生素K依赖性凝血因子II、VII、IX、X及蛋白C/蛋白S的γ-羧基化与合成,产生抗凝作用。特点:①口服有效;②体内有效、体外无效;③起效慢(需数日)、作用时间长;④高度血浆蛋白结合。 |
| 临床应用 | 用于预防与治疗静脉/动脉血栓栓塞性疾病(如深静脉血栓、肺栓塞、心房颤动合并栓塞风险、机械心瓣膜等),需监测INR并个体化给药。 |
| 主要不良反应 | 自发性出血(皮肤、黏膜、内脏出血);早期可出现蛋白C缺乏导致的皮肤坏疽(罕见)。戒断或过量需给维生素K或凝血因子制剂逆转。 |
| 重要药物相互作用(及效应) | 增强抗凝(出血↑): - 广谱抗生素(抑制肠道产维K细菌)→ INR升高(如部分头孢、喹诺酮、甲硝唑等)。 - 肝药酶抑制剂(如甲硝唑、西咪替丁、某些 azole 抗真菌药)→ 代谢↓,抗凝增强。 - 药物位移蛋白结合(阿司匹林、磺胺类、保泰松等)→ 游离药物↑。 - 肝功能不全(肝合成↓)→ 出血风险↑。 减弱抗凝(血栓风险↑): - 肝药酶诱导剂(苯巴比妥、苯妥英、利福平、卡马西平等)→ 代谢加快,INR下降。 - 摄入维生素K或富含维K食物(绿叶菜、大量维K补充)→ 抗凝被拮抗。 监测提示:任何合并用药、肝病、饮食改变或抗生素治疗时应更频繁监测INR并调整剂量。 |
新型口服抗凝药(NOACs)
| 项目 | 要点 |
|---|---|
| 药理作用及机制 | NOACs主要包括:直接凝血酶(IIa)抑制剂:达比加群酯(dabigatran etexilate)和直接Xa因子抑制剂:利伐沙班(rivaroxaban)、阿哌沙班(apixaban)、依度沙班(edoxaban)等。作用机制:选择性、可逆性抑制靶因子→抑制凝血级联中关键酶活性→降低纤维蛋白形成与血栓形成风险。主要临床用途:替代华法林用于非瓣膜性房颤的卒中与系统性栓塞预防,并用于静脉血栓栓塞(VTE)的治疗与预防。 药代学特点:起效快、半衰期较短、与维生素K拮抗剂相比无需常规INR监测,但部分药物受肾功能与P-gp/CYP相互作用影响,需按肾功能与合并用药调整剂量。 |
| 特异性拮抗药 | 发生重大出血或需紧急逆转时:对达比加群可用依达赛珠单抗(idarucizumab)静脉注射快速中和;对Xa抑制剂可选择八因子浓缩物/产物(PCC)或四因子PCC,以及特异性重组因子Xa拮抗剂逆转剂安德沙班(andexanet alfa)(可用时)。逆转策略需参考出血部位、药物最近给药时间、肾功能及可用制剂。 |
抗血小板药
| 项目 | 阿司匹林 | 双嘧达莫 |
|---|---|---|
| 别称 | 乙酰水杨酸 | 潘生丁(dipyridamole) |
| 主要机制 | 抑制血小板聚集(不可逆抑制COX‑1,减少TXA2 产生) | 抑制血小板聚集,体内外均有抗血栓作用(增加细胞内cAMP/影响腺苷代谢) |
| 作用机制(要点) | 1. 不可逆抑制环氧合酶(COX‑1),抑制血小板合成TXA2,降低血小板聚集能力。 2. 对血管内皮PGI2 合成影响小(低剂量主要抑制血小板TXA2)。 3. 大剂量时可抑制内皮PGI2 合成,可能降低PGI2 水平。 | 1. 抑制磷酸二酯酶并阻断腺苷再摄取,使细胞内cAMP 增加,抑制血小板功能。 2. 增强PGI2 活性并促进血管内皮產生PGI2。 3. 轻度抑制血小板环氧合酶/减少TXA2 合成,整体呈抗血栓与血管保护作用。 |
| 临床应用 | 用于预防及治疗动脉血栓性事件:冠心病、心肌梗死二级预防、动脉血栓栓塞防治、缺血性卒中和短暂性脑缺血发作;亦用于深静脉血栓形成与肺梗死的辅助防治(按指征)。 | 用于心血管器械(如心脏瓣膜置换)周围与体外循环场景的抗血栓;用于脑血栓预防与动脉粥样硬化过程干预;常与阿司匹林合用以增强抗血栓效果或在对阿司匹林不耐受者替代/联合应用。 |
H1受体阻断药
- 分类
| 分类 | 代表药物 | 主要特点 | 临床利弊(关键点) |
|---|---|---|---|
| 第一代 H1 受体阻断药 | 苯海拉明、异丙嗪、氯苯那敏 等 | 强进入中枢神经系统、受体选择性差;作用起效快但持续时间短 | 不良:明显镇静(嗜睡)、抗胆碱样作用(口干、眼干、鼻干、便秘、视力模糊) 缺点:耐受性易产生、作用时间短 |
| 第二代 H1 受体阻断药 | 氯雷他定、西替利嗪(仙特敏)、阿司咪唑(息斯敏)等 | 周效或长效,极少进入中枢;受体选择性较高 | 优点:多为长效、无或极少嗜睡 对症:对打喷嚏、流清涕、鼻痒效果好;对鼻塞效果相对较差 |
- 两种常用的药物
| 药物 | 药理作用 | 临床应用 | 不良反应 |
|---|---|---|---|
| 氯苯那敏(第一代 H₁拮抗剂) | A. 阻断外周 H₁受体:可完全对抗组胺引起的支气管、胃肠平滑肌收缩,并抑制组胺介导的毛细血管扩张和通透性增加。 B. 可穿过血脑屏障,产生中枢抑制:表现为镇静、嗜睡;可能因拮抗中枢H₁介导的觉醒反应。 | 主要用于皮肤黏膜过敏性疾病(如荨麻疹、过敏性鼻炎);对昆虫叮咬所致瘙痒/水肿有效;对血清病、药疹、接触性皮炎有一定疗效。 对支气管哮喘疗效差,对过敏性休克无效。 | A. 中枢抑制:镇静、嗜睡、乏力(驾驶员、高空作业者禁用或慎用)。 B. 消化道反应:口干、厌食、便秘或腹泻。 C. 罕见:粒细胞减少、溶血性贫血等。 |
| 氯雷他定(第二代 H₁拮抗剂) | 为阿扎他定衍生物,选择性阻断外周 H₁受体,不穿过血脑屏障(无/极少中枢镇静)且无明显抗胆碱作用;起效快、作用强且持久;可减少IgE介导的组胺释放,单次给药作用可持续约24小时。 | 常用于过敏性鼻炎、慢性荨麻疹及其他过敏性皮肤病,作为首选之一的非镇静抗组胺药。 | 不良反应少见:乏力、头痛、口干;偶见肝功能异常。 哺乳期妇女慎用。 |
作用于呼吸系统与消化系统的药物
平喘药的分类;奥美拉唑的药理作用。
考题
- 【例1】对 β2 受体有选择性激动作用的平喘药是 A. 茶碱 B. 肾上腺素 C. 沙丁胺醇 D. 色甘酸钠 E. 异丙肾上腺素
- 【例2】下列药物中,具有强心作用的药物是 A. 氨茶碱 B. 乙酰唑胺 C. 呋塞米 D. 甘露醇 E. 氢氯噻嗪
- ✨【例3】主要用于预防 I 型变态反应所致哮喘的药物是 A. 氨茶碱 B. 肾上腺素 C. 特布他林 D. 色甘酸钠 E. 异丙肾上腺素
- 【例4】通过抑制 H±K±ATP 酶而用于治疗消化性溃疡的药物是 A. 异丙嗪 B. 肾上腺皮质激素 C. 雷尼替丁 D. 奥美拉唑 E. 苯海拉明
- 【例5】奥美拉唑减少胃酸分泌的机制是可抑制 A. 乳酸脱氢酶 B. Na±K±ATP 酶 C. H±K±ATP 酶 D. HMG-CoA 还原酶 E. 葡萄糖-6-磷酸酶
- 【例6】特异性抑制胃壁细胞质子泵活性的药物是 A. 吡仑西平 B. 奥美拉唑 C. 氢氧化镁 D. 枸橼酸铋钾 E. 雷尼替丁
- 【例7】奥美拉唑的临床应用适应证是 A. 胃肠平滑肌痉挛 B. 萎缩性胃炎 C. 消化道功能紊乱 D. 慢性腹泻 E. 消化性溃疡
基础
平喘药
抗炎平喘药(糖皮质激素)
| 类别 | 要点 | 说明 |
|---|---|---|
| 代表药物 | 常用吸入糖皮质激素(ICS) | 倍氯米松、布地奈德、环索奈德、丙酸氟替卡松(常用制剂多为气雾/粉吸入) |
| 药理作用 | 抑制炎症细胞功能 | 抑制嗜酸细胞、肥大细胞、T细胞、巨噬细胞等参与哮喘的炎症细胞,降低细胞活性与募集 |
| 抑制细胞因子/介质 | 减少IL-4、IL-5、IL-13、TNF等细胞因子及炎症介质的产生,抑制气道炎症级联 | |
| 降低气道高反应性 | 长期应用可降低气道高反应性,减少哮喘发作频率与严重度 | |
| 增强对儿茶酚胺敏感性 | 提高支气管和平滑肌对β-激动剂(儿茶酚胺类)的反应性,协同支气管扩张剂效果 | |
| 临床应用 | 适应证与给药注意 | 用于对支气管扩张剂单用控制不佳的慢性哮喘作为维持/控制治疗;长期吸入可减少发作、降低病情严重度,但不能缓解急性发作。目前多以气雾/粉吸入实现局部给药并减少全身激素用量或替代口服激素。对哮喘持续状态(严重支气管痉挛导致无法有效吸入)吸入制剂疗效受限,应慎用或改用系统给药。 |
| 不良反应 | 常见与少见副作用 | 常规吸入剂量下系统性不良反应少见。局部可见念珠菌性口咽感染、声音嘶哑、咽刺激——使用后漱口或咽部清洁可减少口咽部副作用。长期大剂量或系统吸收增加时可出现生长抑制(儿童)、骨密度下降、肾上腺抑制等系统性效应,应权衡剂量与监测。 |
支气管扩张药
| 类别 / 代表药 | 主要机制(高亮) | 药理作用 | 起效/持续时间 | 临床应用(要点) | 主要不良反应 / 与其它类药区别(要点) |
|---|---|---|---|---|---|
| 选择性 β2 受体激动药 (沙丁胺醇 Salbutamol;特布他林 Terbutaline) | 刺激 β2 受体 → ↑cAMP → 支气管平滑肌松弛 | 快速且强效的支气管扩张;减轻支气管痉挛;吸入优先,系统暴露低 | 吸入:起效 5–15 min(部分制剂 5 min 内);持续 ~3–6 h;口服/肌注起效慢且持续时间视剂型而异 | 一线急性支气管痉挛处置与哮喘缓解剂;喘息型支气管炎 / 支气管痉挛相关呼吸道疾病 | 常见:心悸、震颤、低钾、暂时性血糖升高;高剂量或全身给药可致心率增快和心肌缺血风险;起效快、用于急救 (区别:起效比茶碱快,扩张作用通常强于茶碱) |
| 黄嘌呤类(氨茶碱 / 茶碱 Aminophylline / Theophylline) | 非选择性 PDE 抑制 ↑cAMP/ cGMP;并阻断腺苷受体、可促内源性儿茶酚胺释放 | 缓慢但持久的支气管扩张;中枢兴奋、利尿、强心效果;多靶点作用 | 口服或静脉给药起效慢(口服约30 min 起效,持续数小时);用于维持治疗或当β2 无效时静脉给药 | 慢性哮喘维持治疗(不作为首选)、COPD 改善气促;对中枢型睡眠呼吸暂停有一定作用 | 窄治疗窗:恶心、呕吐、心悸、心律失常、癫痫样癫性发作;与许多药物/疾病相互作用(代谢差异大);与β2 比起效慢且扩张作用弱,但可与之叠加用于难治性发作 |
| 抗胆碱药(异丙托溴铵 Ipratropium;噻托溴铵 Tiotropium) | 阻断 M 胆碱受体(M1–M3),抑制迷走胆碱能引起的支气管收缩 | 通过阻断胆碱能收缩通路引起气道平滑肌松弛;对高迷走张力相关痉挛尤为有效;局部作用为主(吸入) | 异丙托溴铵:吸入,作用 4–6 h;噻托溴铵:长效,单次吸入可持续 ≥24 h(干粉吸入) | 首选或重要药物用于 COPD 维持治疗与缓解支气管痉挛;对不能耐受或对β2 激动药反应差的哮喘患者可用;噻托长期维持 COPD | 局部:口干、咽干;系统吸收少,偶见视物模糊、尿潴留(老年/前列腺肥大需注意);与β2 不同:无 β2 引起的心血管副作用,噻托作用时间长,适合每日一次维持 |
镇咳与祛痰药
- 考得不多
抗酸药(PPI)
| 项目 | 要点 |
|---|---|
| 药理作用与机制 | 1. 抑制胃酸分泌 质子泵抑制剂(PPI)不可逆抑制胃壁细胞H+-K+-ATP酶,使胃酸分泌强力且持久下降。 2. 刺激胃泌素分泌(反馈性) 抑酸后胃内pH升高,G细胞分泌胃泌素增加(临床上与长期PPI使用相关的高胃泌素症需关注)。 3. 胃黏膜保护 奥美拉唑可减少阿司匹林、乙醇、应激相关黏膜损伤的发生,具有一定预防作用。 4. 抗幽门螺杆菌(体外/协同作用) 体外显示抑菌作用,临床上常与抗生素联用以提高根除率(PPI有利于抗生素活性及细菌易位)。 |
| 临床应用 | 消化性溃疡、反流性食管炎、上消化道出血、幽门螺杆菌根除方案的组成部分;亦用于Zollinger–Ellison综合征、长期胃酸抑制需求及与NSAIDs相关黏膜保护等。 |
| 不良反应(常见/重要) | 神经系统 头痛、头晕、失眠,少见外周神经炎。 消化系统 口干、恶心、呕吐、腹胀、便秘或腹泻。 其他/罕见但需关注 男性乳腺发育(长期高剂量/罕见)、皮疹、溶血性贫血;长期PPI相关风险包括低镁血症、骨折风险升高、肠道感染(如C. difficile)及维生素B12吸收降低,临床用药应权衡利弊并定期随访。 |
糖皮质激素类药、抗甲状腺药与降糖药
糖皮质激素的作用及应用;降糖药的作用机制。
考题
- 【例1】关于糖皮质激素抗炎作用的正确叙述是 A. 能提高机体的防御功能 B. 直接杀灭病原体 C. 促进创口愈合 D. 抑制病原菌生长 E. 对抗各种原因,如物理、生物因素等引起的炎症
- 【例2】不属于糖皮质激素类药物抗休克作用机制的是 A. 稳定溶酶体膜 B. 扩张痉挛收缩的血管 C. 抑制炎性细胞因子释放 D. 增强心肌收缩力 E. 中和细菌外毒素
- 【例3】地塞米松的临床应用不包括 A. 风湿性心肌炎 B. 骨质疏松 C. 系统性红斑狼疮 D. 过敏性紫癜 E. 感染中毒性休克
- ✨【例4】糖皮质激素可用于治疗 A. 原发性血小板减少症 B. 急性淋巴细胞白血病 C. 慢性粒细胞白血病 D. 真性红细胞增多症 E. 骨质疏松
- 【例5】长期应用糖皮质激素后,突然停药所产生的反跳现象是由于病人 A. 对糖皮质激素产生耐药性 B. 对糖皮质激素产生了依赖或病情未能完全控制 C. 肾上腺皮质功能亢进 D. 肾上腺皮质功能减退 E. ACTH分泌减少
- 【例6】属于抑制甲状腺激素合成的药物是 A. 放射性碘 B. 酚苄明 C. 左甲状腺素钠 D. 丙硫氧嘧啶 E. 涣隐亭
- 【例7】属于α受体阻断药的是 A. 放射性碘 B. 酚苄明 C. 左甲状腺素钠 D. 丙硫氧嘧啶 E. 涣隐亭
- 【例8】磺酰脲类药物的药理作用为 A. 使电压依赖性钾通道开放 B. 可促进胰岛素释放而降血糖 C. 不改变体内胰高血糖素水平 D. 可使电压依赖性钠通道开放 E. 能抑制抗利尿激素的分泌
基础
糖皮质激素类药(糖皮质激素)
- 药理作用
| 类别 | 主要作用(精简) | 机制要点 |
|---|---|---|
| 治疗性作用 | 抗炎 免疫抑制 抗过敏 抗休克/退热 | 抑制炎症因子产生、抑制淋巴细胞增殖与T细胞介导的细胞效应 减少过敏介质生成;大剂量可改善微循环、稳定溶酶体膜、提高对内毒素的耐受性 |
| 代谢影响 | 升高血糖 加速蛋白分解,抑制合成 脂肪重新分布(中心性肥胖) | 促糖异生(利用蛋白氨基酸合成葡萄糖)、抑制组织对葡萄糖利用;促进蛋白质分解、脂肪向躯干/面部沉积 |
| 水、电解质 | 弱的保钠排钾作用 | 增加肾小球滤过率并对抗利尿激素,促钠潴留、减少肾小管对水的重吸收(长期可致骨质丢失) |
| 血液与造血 | 刺激骨髓造血:RBC、血小板、嗜中性细胞↑ | 减少单核/淋巴细胞外周回流,短期血象表现为中性粒细胞升高与淋巴细胞相对减少 |
| 中枢/精神 | 提高中枢兴奋性,可诱发精神症状 | 剂量或个体差异相关,长期或大剂量可出现失眠、情绪不稳、精神病样表现 |
| 骨骼与心血管 | 骨质疏松(长期) 可致高血压(部分患者) | 抑制成骨、促进骨吸收;促钠潴留与血管敏感性改变引起血压升高 |
- 临床应用
| 适应证 | 要点说明 |
|---|---|
| 严重急性感染 | 用于中毒性或伴休克病例(如中毒性菌痢、中毒性肺炎、暴发型流脑、败血症)。与抗生素合用,作为辅助抗炎和稳定循环的药物。 |
| 抗炎及防治炎症后遗症 | 早期使用可减少炎性渗出,抑制纤维组织过度增生与粘连,降低瘢痕/粘连等后遗症。 |
| 自身免疫性疾病 | 能迅速缓解免疫介导的炎症与症状。如多发性皮肌炎首选药物之一,亦用于系统性红斑狼疮、类风湿关节炎等。 |
| 过敏性疾病 | 对严重或危及脏器的过敏反应可选用,短程控制过敏炎症并防止进展。 |
| 器官移植排斥反应 | 术前或术后用于抑制免疫反应,预防或控制急性排斥。 |
| 抗休克治疗 | 主要用于感染性/中毒性休克的辅助治疗,帮助恢复血管反应性与减轻炎症介导的损伤。 |
| 血液病 | 用于急性淋巴细胞白血病(儿童)、再生障碍性贫血、过敏性紫癜等,改善血象与抑制免疫性破坏。 |
| 局部应用 | 可作外用、局部封闭或关节腔内注射,实现局部高效抗炎并减少系统副作用。 |
| 替代治疗(肾上腺皮质功能不全) | 用于急、慢性肾上腺皮质功能低下的激素替代,维持基础皮质醇水平与应激反应。 |
- 不良反应
| 类别 | 要点 | 临床表现 / 防治要点 |
|---|---|---|
| 长期大剂量应用不良反应 | 内分泌/体形改变 | 满月脸、水牛背、皮肤变薄、多毛、中心性肥胖等。 |
| 感染风险↑ | 诱发新感染或使潜在感染扩散,免疫抑制患者尤甚;出现感染时需及时评估激素用量与抗感染治疗。 | |
| 消化道并发症 | 可诱发或加重消化性溃疡、消化道出血及穿孔;有既往溃疡史者需防治胃黏膜损伤(抑酸、保护粘膜)。 | |
| 心血管/水电解质 | 可致水钠潴留、血压升高、心衰风险增加;监测体重、电解质及血压,必要时利尿/降压治疗。 | |
| 骨骼肌及伤口愈合 | 骨质疏松、肌无力/肌萎缩、伤口愈合延迟;长期者应评估骨密度并采取钙维D及抗骨质疏松治疗方案。 | |
| 代谢:糖代谢异常 | 约50%出现糖耐量受损或糖尿病(类固醇性糖尿病);监测血糖并按需调整降糖治疗。 | |
| 停药反应 | 医源性肾上腺皮质功能不全/危象 | 长期用激素者突然停药(或遇严重应激)可致恶心、呕吐、乏力、低血压、休克。防治:必须缓慢减量;停药后可用ACTH试验评估/停药后连续补用短期糖皮质激素约7天;停药1年内遇应激应给予足量糖皮质激素。 |
| 反跳/病情复发 | 原病未获控制或产生依赖时突然减量/停药可导致症状复发或加重。处理:短期内可能需增量控制病情,症状缓解后再缓慢减量直至停药。 |
甲状腺激素及抗甲状腺药
甲状腺激素
| 项目 | 要点 |
|---|---|
| 药理作用 | 维持正常生长发育 促进代谢与产热 增强交感-肾上腺系统反应性 总体:提高基础代谢率,促进蛋白质、碳水化合物及脂质代谢;对心血管系统有正性变力与变速作用。 |
| 临床应用 | 主要适应证: 1. 甲状腺功能减退(包括先天性或获得性) 2. 单纯性甲状腺肿(弥补甲状腺激素不足以抑制TSH) 3. 用于甲状腺激素替代治疗及抑制TSH的长期管理(剂量应个体化,定期监测TSH、FT4) |
| 不良反应 | 过量或治疗性甲状腺素诱导的表现: 心悸、手抖、多汗、体重减轻、失眠、焦虑 长期过量可致心房颤动、骨密度下降(加速骨吸收)等。 临床提示:对心血管病、骨质疏松患者应谨慎起始剂量并缓慢调整,定期随访TSH/FT4及临床症状。 |
硫脲类
| 项目 | 要点 | 详细描述 |
|---|---|---|
| 代表药物 | 硫脲类抗甲状腺药 | 甲硫氧嘧啶(methimazole)、丙硫氧嘧啶(propylthiouracil, PTU) |
| 药理作用 | 抑制甲状腺激素合成 | 硫脲类抑制甲状腺过氧化物酶,阻断酪氨酸残基的碘化及酪氨酸偶联,减少甲状腺激素(T4、T3)生物合成;对已合成并释放的激素无直接作用。 |
| 抑制外周T4→T3的脱碘 | 丙硫氧嘧啶可抑制外周组织将T4转化为活性T3,能快速降低血清游离T3水平,因此在重症甲亢或甲状腺危象首选PTU。 | |
| 减弱β受体介导的效应 | 硫脲类可减少心肌和骨骼肌的β受体数目并降低腺苷酸环化酶活性,缓解甲亢的交感样症状(非首选替代β阻滞剂的手段,但有辅助作用)。 | |
| 免疫调节作用 | 可降低循环中的甲状腺刺激免疫球蛋白(TSI)水平,具有一定免疫抑制作用,与 Graves 病病情改善相关。 | |
| 临床应用 | 甲亢的一线内科治疗 | 适用于轻—中度甲亢、年轻患者、暂时不适合手术或放射性碘治疗者;推荐疗程通常为1–2年,视病情与复发情况调整。 |
| 术前准备 | 用于基础代谢率较高、需要降代谢与控制甲状腺功能以降低手术风险的患者。 | |
| 甲状腺危象/重症甲亢 | 首选丙硫氧嘧啶(PTU),可使用常规治疗剂量的约2倍短期给药以迅速抑制外周T4→T3的转换,通常疗程不超过1周(同时给予支持治疗、β阻滞剂、糖皮质激素等)。 | |
| 不良反应(常见/严重) | 过敏反应 | 最常见;表现为斑丘疹、皮肤瘙痒、药疹等;出现严重皮疹应停药并评估替代方案。 |
| 胃肠道/味觉改变 | 可有恶心、呕吐、胃肠不适;甲硫氧嘧啶偶见味觉或嗅觉异常。 | |
| 粒细胞缺乏/无粒细胞症 | 最严重的不良反应,通常发生于治疗后2–3个月,表现为发热、咽痛等感染征象,出现应立即停药并检查血常规。 | |
| 甲状腺肿/功能减退 | 长期用药可导致甲状腺肿和甲状腺功能减低,需随访甲功并调整剂量或停药。 |
碘及碘化物
| 条目 | 要点 | 临床要点 |
|---|---|---|
| 1. 药物 | 复方碘溶液、碘化钠、碘化钾(盐中加碘常用碘化钾/碘化钠作添加剂)。 | 剂型:溶液、片剂、加碘食盐。 |
| 2. 药理作用 | 小剂量碘:为甲状腺激素合成原料,能预防单纯性甲状腺肿,早期有效。推荐食盐加碘浓度约按1/100000~1/10000比例加入碘化钾/碘化钠。 | 用途:碘缺乏地区预防、轻度甲状腺肿管理。 |
| 大剂量碘:具抗甲状腺作用,主要抑制甲状腺激素释放并拮抗TSH促释放作用(短期显著)。 | 特点:起效快但多为短暂(Wolff–Chaikoff效应/escape现象需注意)。 | |
| 3. 临床应用 | 术前准备(降低甲状腺血流与激素释放)、甲状腺危象(短期控制甲状腺素释放)。 | 注意:大剂量用于短期控制,长期可导致反跳性甲功变化或缺乏疗效。 |
| 4. 不良反应 | 可见过敏反应(碘过敏)、诱发或加重甲状腺功能紊乱(包括亞急性或自體免疫相关变化)、胃肠不适、皮疹等。 | 临床提示:既往碘敏感史、妊娠晚期、既有甲状腺疾病者用量与疗程需谨慎监测甲功与临床表现。 |
β受体阻断药
| 项目 | 要点 |
|---|---|
| 代表药物 | 普萘洛尔、 metoprolol(美托洛尔)、阿替洛尔 |
| 临床适应证 | 用于甲亢时控制交感症状;适用于不能使用抗甲状腺药物、不能耐受/不宜手术或无法接受放射性碘治疗的甲亢患者作为对症治疗或围术期/急性期稳定心率、减轻震颤与焦虑。 |
| 主要注意/不良反应 | 较少干扰常规甲状腺功能测定与硫脲类药物对甲状腺的作用;但需注意:对心血管(心率过慢、低血压、心力衰竭恶化)及气管平滑肌(支气管痉挛,尤其有哮喘或慢性阻塞性肺病患者)的不良反应,使用时应评估心肺功能并监测生命体征。 |
放射性碘
- 不考
降糖药
用于尿崩症治疗 — 卡马西平、氢噻嗪、噻唑烷二酮、加压素、垂体后叶素。
胰岛素的降糖机制 — 增加葡萄糖的去路,抑制葡萄糖的来源。
罗格列酮的降糖机制 — 为胰岛素增敏剂,增加靶组织对胰岛素的敏感性,改善胰岛素抵抗。
磺酰脲类的降糖机制 — 促进残存胰岛β细胞分泌胰岛素,因此降糖前提是要有β细胞残存。
胰岛素
| 项目 | 要点 | 临床意义 / 备注 |
|---|---|---|
| 药理作用 | 1) 促进合成代谢 - 促进肝脏、脂肪、肌肉的糖原与脂质储存; - 促进糖原合成,抑制糖原分解与糖异生; - 加速葡萄糖氧化与糖酵解,降低血糖。 2) 促进脂肪合成 - 减少游离脂肪酸与酮体生成;增加脂肪酸与葡萄糖转运与利用。 3) 促进蛋白质与核酸合成 - 增加氨基酸转运、蛋白合成,抑制蛋白分解;促进核酸合成。 4) 心血管/肾血流效应 - 增快心率、增强心肌收缩力;可减少肾血流(情境相关)。 | 胰岛素是主要的促合成代谢激素,对降低血糖、抑制酮体生成及维持能量代谢关键。治疗中需平衡降糖与低血糖风险。 |
| 临床应用 | 1) 胰岛素注射剂 主要用于: - 1型糖尿病(必需); - 2型糖尿病:口服药物控制不佳、发生急性/严重并发症、应激状态(感染、手术等)、伴有细胞内缺钾时。 2) 胰岛素吸入剂 - 可减轻长期反复皮下注射的痛苦,改善生活质量(适应证与禁忌依制剂不同)。 | 选择制剂(速效、常规、中效、长效)与给药方案应根据血糖谱、生活方式与并发症调整;吸入制剂注意呼吸系统禁忌。 |
| 不良反应 | 1) 低血糖 — 最常见且最严重;表现:出汗、心悸、震颤、意识障碍,严重可致昏迷。 2) 过敏反应 — 罕见,局部或全身性。 3) 胰岛素抵抗 — 急性:常因感染、创伤、手术等应激;慢性:病因复杂(抗体、肥胖、药物等)。 4) 注射部位反应 — 脂肪萎缩或增生(脂肪萎缩常见于女性)。 | 管理要点:监测血糖与低血糖风险、轮换注射部位、评估抵抗原因、对过敏/严重低血糖准备应急方案(葡萄糖、胰高血糖素等)。 |
其它降糖药
| 代表性药物 | 药理作用 | 临床应用 | 不良反应 |
|---|---|---|---|
| 二甲双胍、苯乙双胍 | 降低肝糖异生;促进外周(尤其脂肪、肌肉)对葡萄糖摄取;减少肠道葡萄糖吸收;抑制胰高血糖素释放。 | 首选或早期治疗2型糖尿病,适用于肥胖或单用饮食控制无效者(注意:苯乙双胍因乳酸性酸中毒风险多被停用或限制使用)。 | 食欲下降、恶心、腹泻;乳酸性酸中毒(罕见但严重,肾功能受损或低灌注时风险↑);维生素B12吸收减少。 |
| 甲苯磺丁脲、格列本脲、格列吡嗪等(磺酰脲类) | 刺激胰岛β细胞释放胰岛素;可降低血糖并增强胰岛素与靶组织结合性。 | 2型糖尿病伴有剩余胰岛功能、饮食控制无效者;(其他特殊适应证如氯磺丙脲在特定非糖尿病适应证上曾用)。 | 低血糖(常见且严重)、体重增加;皮疹、胃肠不适、嗜睡、罕见肝损害、黃疸。 |
| 阿卡波糖(α-葡萄糖苷酶抑制剂) | 抑制小肠刷状缘α-糖苷酶,延缓寡糖/双糖水解及葡萄糖生成,减慢餐后葡萄糖吸收。 | 以餐后高血糖为主的2型糖尿病,可与其他药物联合使用。 | 主要为胃肠道反应:腹胀、放气增多、腹泻、腹痛;少见低血糖(合用胰岛素或促泌剂时)。 |
| 吡格列酮、罗格列酮(胰岛素增敏剂/噻唑烷二酮类) | 激动PPARγ,改善胰岛素抵抗、调节脂质代谢并改善胰岛β细胞功能。 | 2型糖尿病,尤其伴胰岛素抵抗者,可与其他降糖药联合。 | 液体潴留/水肿、体重增加;心衰风险增加(有心衰史慎用);骨折风险增加;肝功能监测需注意。 |
| GLP-1 受体激动剂(例:依克那肽/外泌肽类) | 模拟GLP-1:增强餐后葡萄β细胞依赖性胰岛素分泌;抑制胰高血糖素;延缓胃排空并可降低食欲。 | 2型糖尿病(有体重控制需求者优选,可降低心血管事件风险的药物选择视具体制剂与证据而定)。 | 胃肠道反应(恶心、呕吐、腹泻、食欲下降);少见胰腺炎报告;注射部位反应。 |
| DPP-4 抑制剂(西他列汀、维格列汀) | 抑制DPP-4酶,减少内源性GLP-1和GIP降解,增强葡萄依赖性胰岛素分泌并抑制胰高血糖素释放。 | 2型糖尿病,常与其他口服药或胰岛素联用,低血糖风险较促泌剂低。 | 通常耐受性好:可能发生上呼吸道感染、头痛、少数报道胰腺炎、罕见关节痛或皮疹;个别报告免疫相关不良反应。 |
子宫平滑肌兴奋药
- 少考
β-内酰胺类、大环内酯类与林可霉素类抗生素
各类抗生素的抗菌谱。
考题
- 【例1】对青霉素G最敏感的病原体是 A. 立克次体 B. 钩端螺旋体 C. 衣原体 D. 支原体 E. 真菌
- 【例2】青霉素对革兰氏阳性(G+)菌作用的机制是 A. 干扰细菌蛋白质合成 B. 抑制细菌核酸代谢 C. 抑制细菌脂代谢 D. 破坏细菌细胞膜结构 E. 抑制细菌细胞壁肽聚糖(黏糖)的合成
- 【例3】青霉素G的主要不良反应是 A. 肾损害 B. 过敏 C. 听力减退 D. 肝损害 E. 胃肠道反应
- 【例4】β-内酰胺类的作用机制是 A. 抑制细菌细胞壁合成 B. 抑制细菌蛋白质合成 C. 抑制细菌DNA合成 D. 抑制细菌二氢叶酸还原酶 E. 抑制细菌DNA依赖的RNA聚合酶
- 【例5】喹诺酮类的作用机制是 A. 抑制细菌细胞壁合成 B. 抑制细菌蛋白质合成 C. 抑制细菌DNA合成 D. 抑制细菌二氢叶酸还原酶 E. 抑制细菌DNA依赖的RNA聚合酶
- 【例6】第三代头孢菌素的作用特点是 A. 主要用于轻、中度呼吸道和尿路感染 B. 对革兰氏阴性菌有较强的作用 C. 对组织穿透力弱 D. 对肾脏毒性较第一、第二代头孢菌素大 E. 对β-内酰胺酶的稳定性较第一、第二代头孢菌素低
- 【例7】女,32岁。发热、腰痛、尿频、尿急1个月,近3天全身关节酸痛、尿频、尿急加重。体检:体温39.5℃,白细胞13×10^9/L,中性粒细胞86%,尿培养大肠埃希氏菌阳性,诊断为大肠埃希氏菌性尿路感染,应首选 A. 青霉素 B. 红霉素 C. 氟喹诺酮类 D. 头孢曲松 E. 林可霉素
- 【例10】红霉素主要用于治疗的感染 A. 真菌感染 B. 结核分枝杆菌感染 C. 肠道寄生虫感染 D. 肺炎链球菌感染 E. 肠道革兰氏阴性杆菌感染
- 【例11】阿米卡星主要用于治疗的感染为 A. 真菌感染 B. 结核分枝杆菌感染 C. 肠道寄生虫感染 D. 肺炎链球菌感染 E. 肠道革兰氏阴性杆菌感染
- 【例12】阿昔洛韦 A. 具有较强抗铜绿假单胞菌作用 B. 主要用于金黄色葡萄球菌引起的骨与关节感染 C. 为支原体肺炎首选药物 D. 具有抗DNA病毒的作用 E. 对念珠菌有强大抗菌作用
- 【例13】克林霉素 A. 具有较强抗铜绿假单胞菌作用 B. 主要用于金黄色葡萄球菌引起的骨与关节感染 C. 为支原体肺炎首选药物 D. 具有抗DNA病毒的作用 E. 对念珠菌有强大抗菌作用
基础
β-内酰胺类
| 类型 | 敏感菌 | 临床应用 | 不良反应 |
|---|---|---|---|
| 窄谱青霉素类 (青霉素 G) | 高度敏感:G+ 球菌(溶血性链球菌、肺炎球菌、草绿色链球菌、部分金黄色葡萄球菌、表皮葡萄球菌); 部分 G- 杆菌、螺旋体(梅毒螺旋体)及放线菌等 | 首选用于由敏感菌致的感染: 咽炎、肺炎、皮肤软组织感染、脑膜炎、败血症、心内膜炎、梅毒、炭疽等 | ① 变态反应(最常见,严重可致过敏性休克); ② 赫氏反应(治疗梅毒等时短期症状加重:发热、寒战、心动过速等); ③ 肌注部位局部反应(疼痛、红肿、硬结)。 |
| 广谱青霉素类 (氨苄西林 Ampicillin) | 敏感:G- 杆菌如伤寒沙门菌、副伤寒沙门菌、百日咳鲍特菌、某些大肠埃希菌及变形杆菌;球菌如链球菌; 对耐药金黄色葡萄球菌常无效 | 用于敏感菌所致的呼吸道、泌尿生殖道、胃肠道、软组织、脑膜炎、败血症等感染;可用于伤寒、副伤寒等 | 常见:过敏反应(同青霉素类); 胃肠道不良反应、皮疹、偶发血液学或肝功能异常; 对金黄葡萄球菌耐药。 |
| 广谱青霉素类(口服) (阿莫西林 Amoxicillin) | 与氨苄西林相似,敏感:呼吸道及某些肠道致病菌;对肺炎球菌、奈瑟球菌、沙门菌属、幽门螺杆菌等有较强作用 | 用于呼吸道、泌尿道、胆道感染、伤寒、慢性活动性胃炎及消化性溃疡(与抑酸剂/质子泵抑制剂联合用于幽门螺杆菌根除)等由敏感菌引起的感染 | 常见:过敏反应(包括皮疹、严重者休克); 胃肠道症状、皮疹、罕见血液或肝功能异常; 与氨苄西林类似的不良反应谱。 |
头孢菌素类
| 代 | 代表药物 | 抗菌谱(要点) | 对β-内酰胺酶稳定性 | 临床主要应用 |
|---|---|---|---|---|
| 第一代 | 头孢唑啉、头孢唑肟 | 主要对革兰阳性菌有效(对葡萄球菌、链球菌强);对革兰阴性菌作用弱 | 可被多数β-内酰胺酶破坏(稳定性较差) | 敏感菌所致的皮肤软组织、呼吸道、尿路等感染 |
| 第二代 | 头孢呋辛、头孢孟多(头孢孟多西) | 对革兰阳性仍有效,较第一代稍弱;对部分革兰阴性菌(如肺炎球菌、流感嗜血杆菌)增强;对铜绿假单胞菌无效 | 对部分β-内酰胺酶有一定稳定性(比第一代强) | 敏感菌致的肺炎、胆道感染、尿路感染及其他组织器官感染 |
| 第三代 | 头孢噻肟、头孢曲松 | 对革兰阴性菌作用显著增强(对某些革兰阳性较第二代弱);对厌氧菌也有一定强化 | 对β-内酰胺酶较稳定(稳定性良好) | 危及生命的败血症、脑膜炎、严重肺炎、骨髓炎、复杂尿路感染及铜绿假单胞菌以外的重症感染 |
| 第四代 | 头孢吡罗、头孢匹罗(头孢吡罗烯/头孢匹罗) | 兼具对革兰阳性和革兰阴性菌的强效作用,含对某些耐药革兰阴性菌活性较高 | 对β-内酰胺酶高度稳定(稳定性最高) | 用于对第三代耐药或严重革兰阴性菌感染的治疗(需根据菌谱和耐药性指导使用) |
碳青霉烯类、β-内酰胺酶抑制药
- 优立新(氨苄西林+舒巴坦)、奥格门汀(阿莫西林+克拉维酸)。
| 类别 | 代表药物 | 抗菌谱(要点) | 临床应用(要点) |
|---|---|---|---|
| 碳青霉烯类 | 亚胺培南(imipenem)、美罗培南(meropenem) | 广谱抗菌:对多数革兰阳性(含部分耐药株)、革兰阴性需氧菌和厌氧菌均有良好活性;对产ESBL的肠杆菌效果好;对部分耐药铜绿假单胞菌和某些耐药革兰阳性菌活性视药物而异。 | 用于严重或复杂感染:败血症、复杂腹腔感染、下呼吸道感染(包括医院获得性)、尿路感染、皮肤/软组织感染、骨髓炎等,尤其适用于疑有多重耐药菌或需广谱覆盖时。 |
| β-内酰胺酶抑制剂 | 克拉维酸(clavulanic acid)、舒巴坦(sulbactam)(常与青霉素类合用) | 作用并非直接广谱杀菌,而是抑制β-内酰胺酶,从而恢复伴用青霉素/头孢对产酶菌株的活性。能增强对常见产酶革兰阳性与革兰阴性菌(如部分金黄色葡萄球菌、肠杆菌科、流感嗜血杆菌、拟杆菌等)的疗效;对铜绿假单胞菌、某些沙门氏菌及使用非酶性耐药机制(外排泵、靶位改变)的菌无效或效果差。舒巴坦对阿克曼菌(Acinetobacter)有一定自身活性。 | 与合适的β‑内酰胺类联合用于由产β‑内酰胺酶菌株引起的各类感染:呼吸道、泌尿道、皮肤软组织、腹腔及妇科感染等;用于扩展抗菌谱以覆盖疑为产酶菌的感染。 |
大环内酯、林可霉素、糖肽类
| 类型 | 代表药物 | 机制 | 临床应用 | 不良反应 |
|---|---|---|---|---|
| 大环内酯类 | 红霉素(代表) | 不可逆结合细菌核糖体50S亚基,抑制肽链延长,阻断蛋白质合成。 | 适用于青霉素过敏者及耐青霉素或对青霉素疗效不佳的革兰阳性菌感染(如金黄色葡萄球菌、链球菌);对部分革兰阴性菌(脑膜炎奈瑟菌、淋病奈瑟菌、流感嗜血杆菌、军团菌)和非典型病原体(肺炎支原体、衣原体、立克次体、螺旋体)有效;用于口腔厌氧感染及呼吸道、泌尿生殖系非典型病原体感染。 | 胃肠道反应(常见)、肝损害、过敏性皮疹、药物热、耳鸣(高剂量或长期用药)。 |
| 林可酰胺类 | 林可霉素、克林霉素(常用) | 不可逆结合细菌核糖体50S亚基,阻断肽链延长并抑制蛋白质合成(克林霉素活性较强)。 | 首选药用于金黄色葡萄球菌所致骨髓炎;对各类厌氧菌(包括脆弱拟杆菌)有效,用于口腔、腹腔、妇科感染及厌氧性脓肿;也用于需氧革兰阳性菌引起的呼吸道、骨软组织、胆道感染、败血症、心内膜炎等。 | 胃肠道反应、伪膜性肠炎(克林霉素相关风险↑)、过敏反应、黄疸、肝功能损害。 |
| 糖肽类(多肽类) | 万古霉素(代表) | 与细胞壁前体肽(D-Ala-D-Ala)结合,阻断肽聚糖交联与细胞壁合成,导致细胞裂解(细胞壁合成抑制,杀菌)。 | 用于严重的革兰阳性菌感染,尤其对耐甲氧西林金黄色葡萄球菌(MRSA)、耐甲氧西林表皮葡萄球菌(MRSE)及肠球菌所致败血症、心内膜炎、骨髓炎、重症呼吸道感染等。 | 耳毒性、肾毒性、输注相关反应(“红人综合征”)、过敏反应。 |
氨基糖苷类、四环素类及氯霉素类抗生素
考点散乱。
考题
- 【例1】氨基糖苷类抗生素的抗菌机制是 A. 抑制细菌蛋白质合成 B. 抑制细菌细胞壁合成 C. 影响细菌细胞膜通透性 D. 抑制细菌 RNA 合成 E. 抑制细菌 DNA 合成
- 【例2】链霉素和红霉素抗菌作用针对的细菌结构部位是 A. 细胞壁上肽聚糖 B. 细胞壁上脂多糖 C. 细胞质中核糖体 D. 细胞膜上中介体 E. 细胞染色体 DNA
- 【例3】氨基糖苷类抗生素的主要不良反应是 A. 抑制骨髓 B. 耳毒性 C. 肝毒性 D. 心脏毒性 E. 消化道反应
- 【例4】对铜绿假单胞菌作用最强的氨基糖苷类抗生素是 A. 卡那霉素 B. 庆大霉素 C. 阿米卡星 D. 安布霉素 E. 链霉素
- 【例5】对治疗立克次体感染最有效的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例6】能有效控制铜绿假单胞菌感染的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例7】能抑制 DNA 病毒的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例8】多西环素的药理作用是 A. 对病毒感染有效 B. 对念珠菌属感染有效 C. 杀灭结核分枝杆菌 D. 抑制二氢蝶酸合酶活性 E. 对立克次体感染有效
- 【例9】磺胺药的药理作用是 A. 对病毒感染有效 B. 对念珠菌属感染有效 C. 杀灭结核分枝杆菌 D. 抑制二氢蝶酸合酶活性 E. 对立克次体感染有效
基础
氨基糖苷类抗生素
对铜绿假单胞菌最敏感的氨基糖苷类抗生素是妥布霉素,
对铜绿假单胞菌最敏感的喹诺酮类杭生素是环丙沙星。
抗菌谱最广的氨基糖苷类是阿米卡星。
严重G-杆菌感染首选的氨基糖苷类是庆大霉素。
- 基本知识点
| 项目 | 要点 | 说明(关键点已高亮) |
|---|---|---|
| 抗菌谱 | 需氧G‑杆菌优效 对G球菌活性较差 对肠球菌/厌氧菌不敏感 | 对多种需氧革兰氏阴性杆菌:大肠埃希菌、铜绿假单胞菌、变形杆菌、克雷伯菌、肠杆菌、志贺菌、枸橡酸杆菌等;对革兰氏阳性球菌(如淋病/脑膜炎奈瑟菌)活性较差;对产碱杆菌、不动杆菌、嗜血杆菌、沙门菌有一定作用。对肠球菌与厌氧菌通常不敏感。对部分耐甲氧西林葡萄球菌(MRSA、MRSE)可有活性(视具体药物)。链霉素、卡那霉素对结核分枝杆菌有效。 |
| 抗菌机制 | 主要机制 | 抑制细菌蛋白质合成并破坏细胞膜完整性。包括:①抑制70S起动复合物形成;②与30S亚基结合,导致mRNA错译产生异常蛋白;③阻滞肽链释放使肽链不能释放;④抑制70S解离,阻碍核糖体循环利用。 |
| 膜损伤 | 破坏细菌胞质膜通透性,增强药物内流并加速细胞死亡(与蛋白合成抑制协同)。 | |
| 耐药相关 | 常见机制:酶修饰(磷酸化、腺苷化、乙酰化)、靶位点(核糖体)改变、透入口径/外膜改变和主动外排。 | |
| 临床意义 | 需根据药敏结果选择,结合剂量/给药方式(静脉、肌肉、局部)以避免毒性并克服耐药。 | |
| 临床适应证 | 系统性需氧G‑杆菌感染为主 | 用于敏感需氧革兰氏阴性杆菌所致的全身感染:脑膜炎、呼吸道、泌尿道、皮肤软组织、胃肠道、创伤、骨关节等;常与β‑内酰胺类联合以扩大谱和协同杀菌。 |
| 主要不良反应 | 耳毒性(重要) | 前庭与耳蜗神经损伤:前庭损伤表现为眩晕、眼震、共济失调;耳蜗损伤表现为耳鸣、进行性听力下降至不可逆聋。胎儿内用药亦可致听力损害。不同药物发生率有差异(从高到低大致:neomycin、kanamycin、streptomycin、sisomicin、amikacin、gentamicin、tobramycin、netilmicin、etimicin/isepamicin等)。 |
| 肾毒性 | 肾小管毒性为主,可能致药源性急性肾损伤。发生率受剂量、用药持续时间、合并用药(如环丙沙星、利尿剂)、既往肾功能及年龄影响。个体药物肾毒性发生率排序亦有差异(部分药物高于他药)。 | |
| 神经肌肉阻滞 | 可引起呼吸抑制、肌无力,因阻断乙酰胆碱释放/作用,特别在合并肌松药、重症肌无力或高浓度时风险增加。 | |
| 过敏反应 | 包括皮疹、发热、血管神经性水肿,局部用药(如新霉素)可引起接触性皮炎;链霉素可罕见诱发过敏性休克。 | |
| 用药与监测要点 | 监测血药浓度、肾功能与听力 | 给药前评估肾功能与基线听力/前庭功能;对长期或高剂量治疗监测血药浓度(谷浓度尤重要)、血肌酐、尿量;出现听力/平衡异常应立即停药并评估药物替代方案。 |
- 常用药物
| 庆大霉素 (Gentamicin) | 妥布霉素 (Tobramycin) | 阿米卡星 (Amikacin) |
|---|---|---|
| 给药 口服难吸收,常肌内注射/静脉给药 | 给药 口服难吸收,常肌内注射/静脉给药 | 给药 主要肌内或静脉注射 |
| 临床应用 1. 首选治疗G-杆菌感染(对沙雷菌属作用较强) 2. 与青霉素合用:治疗肺炎球菌、铜绿假单胞菌、肠球菌、葡萄球菌、草绿色链球菌等感染 3. 常用于术前预防和术后感染 | 临床应用 1. 抗菌谱与庆大霉素相似,对肺炎杆菌、肠杆菌属、变形杆菌属的抑菌/杀菌作用比庆大霉素强约4倍和2倍(分别) 2. 对铜绿假单胞菌作用约为庆大霉素的2–5倍 3. 对其他G-杆菌的抗菌活性不如庆大霉素(总体不良反应较庆大霉素轻) | 临床应用 1. 抗菌谱较广,对多数G-杆菌及金黄色葡萄球菌均有较强作用,但对庆大霉素敏感性较弱 2. 对某些氨基糖苷耐药菌仍有效,常作为耐药或重症感染时首选 3. 用于治疗粒细胞减少或免疫缺陷者严重G-杆菌感染 |
| 不良反应 - 耳毒性、肾毒性 - 神经肌肉阻滞、过敏反应 | 不良反应 - 总体不良反应较庆大霉素轻(耳/肾毒性较轻) | 不良反应 - 耳毒性强于庆大霉素 - 肾毒性低于庆大霉素 |
四环素类、氯霉素类
临床上现在已经较少使用
| 抗生素 | 机制 | 临床应用 | 不良反应 |
|---|---|---|---|
| 四环素 | 与细菌核糖体30S 亚基结合,抑制肽链延长与蛋白质合成;主要为抑菌作用。 对革兰阴性菌抑制一般优于革兰阳性菌(但对某些革兰阳性菌活性不及青霉素/头孢)。 | 因耐药增加及副作用,通常非首选。仍用于支原体肺炎、衣原体感染及某些敏感的非典型/特殊感染。 | 局部刺激、继发感染;对儿童可抑制骨骼与牙齿发育(着色);长期或广谱应用可致菌群失调。 |
| 氯霉素 | 与细菌核糖体50S 亚基结合,抑制蛋白质合成,属宽谱抑菌药,对部分细菌(如流感嗜血杆菌、脑膜炎奈瑟菌、肺炎链球菌)可有杀菌作用。 | 因严重毒性已少用,仅在对其他药物无效或忌用时权衡使用(如某些脑膜炎或严重感染的替代方案)。 使用必须严格把握适应证。 | 主要为血液系统毒性(可致不可逆/可逆的再生障碍性贫血或严重粒细胞减少),新生儿可发生灰婴综合征;并可有胃肠道反应。 |
人工合成的抗菌药、抗病毒药与抗真菌药
氟喹诺酮类的作用机制;磺胺类的作用机制; 抗病毒药和抗真菌药的抗菌谱。
考题
糖蛋酮D核胺(核酸-磺胺;和平安康)氨基糖苷类的抗菌机制是抑制细菌蛋白质合成。
第三代喹诺酮类的抗菌机制是抑制细菌 DNA 合成。
磺胺类的抗菌机制是抑制细菌核酸合成。
青霉素类的抗菌机制是抑制细菌细胞壁的合成。
- 【例1】第三代喹诺酮类药物的抗菌机制是其抑制了细菌的 A. 蛋白质合成 B. 细胞壁合成 C. DNA 螺旋酶
- ✨【例2】不属于氟喹诺酮类药物药理学特性的是 A. 抗菌谱广 B. 口服吸收好 C. 与其他抗菌药物无交叉耐药性 D. 不良反应较多 E. 体内分布较广
- 【例3】孕妇,25岁,孕37周。检查发现小阴唇内侧小菜花状赘生物,同时合并肺部感染。针对该患者抗感染治疗,不能使用的药物是 A. 红霉素 B. 喹诺酮类 C. 头孢菌素类 D. β-内酰胺类 E. 青霉素类
- 【例4】女,16岁。随旅行团到偏远地区旅游,晚饭后发生腹痛、腹泻。1天腹泻4次,于当地卫生院治疗后好转。第2天烈日下阳光照射后皮肤出现红斑、瘙痒。导致该不良反应的药物可能是 A. 头孢他定 B. 庆大霉素 C. 盐酸小檗碱 D. 氟氯沙星 E. 吡嗪酰胺
- 【例5】磺胺类药物的抗菌机制主要是抑制 A. 二氢蝶酸合成酶 B. 二氢叶酸还原酶 C. 四氢叶酸合酶 D. DNA 回旋酶 E. 二氢蝶酸合成酶+二氢叶酸还原酶
- 【例6】甲氧苄啶的抗菌机制主要是抑制 A. 二氢蝶酸合成酶 B. 二氢叶酸还原酶 C. 四氢叶酸合酶 D. DNA 回旋酶 E. 二氢蝶酸合成酶+二氢叶酸还原酶
- 【例7】复方磺胺甲噁唑的抗菌机制主要是抑制 A. 二氢蝶酸合成酶 B. 二氢叶酸还原酶 C. 四氢叶酸合酶 D. DNA 回旋酶 E. 二氢蝶酸合成酶+二氢叶酸还原酶
- 【例8】对立克次体感染最有效的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例9】能有效控制铜绿假单胞菌感染的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例10】能抑制 DNA 病毒的药物是 A. 四环素 B. 利巴韦林 C. 妥布霉素 D. 氟康唑 E. 林可霉素
- 【例11】阿昔洛韦 A. 具有较强抗铜绿假单胞菌作用 B. 主要用于金黄色葡萄球菌引起的骨与关节感染 C. 为支原体肺炎首选药物 D. 具有抗 DNA 病毒的作用 E. 对念珠菌有强大抗菌作用
- 【例12】克林霉素 A. 具有较强抗铜绿假单胞菌作用 B. 主要用于金黄色葡萄球菌引起的骨与关节感染 C. 为支原体肺炎首选药物 D. 具有抗 DNA 病毒的作用 E. 对念珠菌有强大抗菌作用
- 【例13】女,32岁。恶心、头晕、呕吐5天。查体:T 38.9℃,R 16次/分,P 80次/分,BP 128/80 mmHg。心、肺(-)。脑膜刺激征阳性。实验室检查:外周血 WBC 异常,HIV 抗体阳性。脑脊液:细胞数(200~300)×10^6/L,墨汁染色(+)。患者的治疗药物应选择 A. 伊曲康唑 B. 氟康唑 C. 伏立康唑 D. 恩替卡韦 E. 两性霉素
- 【例14】属于广谱抗真菌药物的是 A. 利福平 B. 利巴韦林 C. 伯氨喹 D. 氟康唑 E. 环磷酰胺
- 【例15】用于治疗麻风病的药物是 A. 利福平 B. 利巴韦林 C. 伯氨喹 D. 氟康唑 E. 环磷酰胺
- 【例16】用于器官移植排斥反应的药物是 A. 利福平 B. 利巴韦林 C. 伯氨喹 D. 氟康唑 E. 环磷酰胺
基础
喹诺酮类抗菌药
| 项目 | 要点 |
|---|---|
| 代表药物 | 氟喹诺酮类(第三代喹诺酮/氟喹诺酮):诺氟沙星、环丙沙星、氧氟沙星、左氧氟沙星等。 |
| 抗菌机制 | 主要靶点: DNA 回旋酶(DNA gyrase) 和 拓扑异构酶 IV。通过与酶‑DNA 形成三元复合物,抑制切口/封口活性,阻断细菌 DNA 复制,产生活性杀菌效应;对 G+ 与 G- 的作用靶点侧重不同(gyrase 在 G- 中更重要,topo IV 在 G+ 中更重要)。 |
| 抗菌谱与作用 | 广谱杀菌:对多数革兰氏阴性菌、部分革兰氏阳性菌有效;对结核分枝杆菌、军团菌、支原体、衣原体有活性。对厌氧菌(如脆弱类杆菌、梭杆菌等)亦常有较强活性。环丙沙星对铜绿假单胞菌杀灭活性较强。 |
| 临床应用 | 用于对氟喹诺酮敏感菌所致感染:泌尿生殖系统感染、呼吸道感染、肠道感染及其他部位感染(按药物谱及当地耐药情况选择)。 |
| 不良反应(主要) | ① 胃肠道:胃部不适、恶心、呕吐、腹泻,多为轻‑中度,可耐受。 ② 中枢神经:轻者失眠、头晕、头痛;重者可出现精神异常、抽搐、惊厥。发生机制与抑制 GABA 受体并激动 NMDA 受体有关,有癫痫或精神病史者慎用/禁用。 ③ 光敏反应:光照部位瘙痒性红斑,重者可发生皮肤糜烂脱落。 ④ 心脏毒性:可致 QT 间期延长、尖端扭转型室速、室颤等,虽罕见但严重。 ⑤ 软骨/骨骼:影响胎儿及生长期儿童软骨发育,禁用于孕妇及骨骼未发育完全的儿童。 |
磺胺类抗菌药
恶(噁)合成,甲氧还原
| 类别 | 要点 |
|---|---|
| 代表药物 | 柳磺吡啶、磺胺嘧啶、磺胺异恶唑、磺胺甲噁唑(SMZ)等;联合制剂常见为 SMZ + 甲氧苄啶(TMP,trimethoprim)。 |
| 抗菌机制 | 细菌需自身合成叶酸,以PABA为底物:PABA →(二氢蝶酸合成酶/DHPS)→ 二氢蝶酸 →(二氢叶酸还原酶/DHFR)→ 四氢叶酸(辅酶,参与嘧啶/嘌呤合成)。 磺胺类药物与PABA竞争二氢蝶酸合成酶(DHPS),阻断二氢蝶酸合成;TMP抑制二氢叶酸还原酶(DHFR),✨两者联用呈协同杀菌(阻断叶酸代谢上、下游)。哺乳动物可利用外源叶酸,故正常宿主细胞核酸合成不受影响。 |
| 抗菌谱 | 对多数革兰阳性和革兰阴性菌均有活性,典型敏感菌包括A组链球菌、肺炎链球菌、脑膜炎奈瑟菌、淋病奈瑟菌、鼠疫耶尔森菌等;对衣原体(沙眼衣原体)、疟原虫、卡氏肺孢子虫(Pneumocystis jirovecii)、弓形虫滋养体有抑制作用。对支原体、立克次体、螺旋体通常无效。某些制剂(如磺胺米隆、磺胺嘧啶银)对铜绿假单胞菌有活性。 |
| 临床应用 | 用于敏感菌感染,但因不良反应而受限。联合SMZ‑TMP常用于:泌尿道感染、肺孢子虫肺炎(PCP)预防/治疗、弓形虫感染、部分肠道/软组织感染及在暴发性脑脊髓膜炎、鼠疫等历史性疗效显著的感染中仍有应用。注意耐药性与地区差异。 |
| 主要不良反应 | ① 泌尿系:结晶尿/尿路梗阻、肾损害(尤在脱水或高剂量下);② 过敏反应:皮疹、发热,存在交叉过敏,严重者可出现Stevens‑Johnson综合征/中毒性表皮坏死松解(罕见);③ 血液系统:可抑制骨髓导致粒细胞减少、再生障碍性贫血、血小板减少;④ 神经系统:头晕、头痛、乏力、失眠(少数)——用药期间避免高空作业/驾驶;⑤ 其他:恶心、呕吐、上腹不适、食欲下降。用药监测:肾功能、血象、过敏反应。禁用/慎用于对磺胺过敏者及部分妊娠/新生儿情况(黄疸风险)。 |
甲氧苄啶(TMP)
| 项目 | 要点 |
|---|---|
| 机制 | 甲氧苄啶(TMP):抑制细菌二氢叶酸还原酶(DHFR),阻断四氢叶酸合成,属于抑菌药。 单用时与磺胺类药物均为抑菌;与SMZ合用时呈协同抑菌。 |
| 抗菌谱与活性 | 抗菌谱与磺胺甲噁唑(SMZ)大致相近;TMP对DHFR的抑制活性显著强于SMZ(常为数十倍)。 单独使用为抑菌;两药按5:1(SMZ:TMP)配伍为常用复方制剂以扩大疗效并减少耐药。 |
| 临床适应证 | 常用复方:复方磺胺甲噁唑(SMZ‑TMP,5:1,复方新诺明)。 主要适应证包括: - 泌尿道感染:大肠埃希菌、变形杆菌、克雷伯菌引起的UTI。 - 上/下呼吸道感染:肺炎链球菌、流感嗜血杆菌等(视耐药谱决定)。 - 肠道感染:志贺菌属、伤寒沙门菌、霍乱弧菌等(根据当地耐药性和指征)。 - 皮肤/软组织及特定感染:卡氏肺孢子虫肺炎(Pneumocystis jirovecii)的预防与治疗、诺卡菌属(Nocardia)感染、腹股沟肉芽肿(肉芽肿性淋病/Donovanosis)等。 |
| 主要不良反应 | 可引起叶酸代谢受损,导致巨幼细胞性贫血、白细胞减少、血小板减少;其他常见/重要不良反应包括: - 过敏反应(皮疹、严重者可发生Stevens‑Johnson综合征/中毒性表皮坏死溶解)。 - TMP特有:高钾血症(尤其合并ACEI/ARBs或肾功能不全者)。 - SMZ特有:新生儿胆红素脑病(禁用于1个月内新生儿)。 - 其他:肝功能异常、肾结晶/尿路症状、光敏反应等。 |
甲硝唑
| 项目 | 要点 | 详细说明 |
|---|---|---|
| 抗菌机制 | 硝基还原→损伤DNA | 甲硝唑为硝基咪唑类,在细胞内低氧/无氧条件下其分子硝基被还原为活性中间体(最终可达氨基),这些还原产物与病原体DNA反应并引起链断裂与合成抑制,导致细菌/原虫死亡。 |
| 抗菌谱 | 主要抗厌氧菌与原虫 | 对厌氧细菌(如脆弱类杆菌)高度敏感;对原虫(滴虫、阿米巴滋养体)和某些厌氧革兰阳性梭菌(如破伤风梭菌)有强杀灭作用。 对需氧菌或兼性需氧菌通常无效。 |
| 临床适应证 | 厌氧感染与特定原虫病首选 | 用于厌氧菌引起的口腔、腹腔、妇科(生殖道)、下呼吸道、骨关节感染;对幽门螺杆菌相关消化性溃疡(常与其他药物联合)、对四环素耐药之艰难梭菌所致假膜性肠炎有疗效;为阿米巴病、滴虫病与破伤风治疗的首选或常用药。 |
| 局限与注意事项 | 需氧环境无效;警惕毒副作用与药物相互作用 | 在需氧菌感染单独使用无效;常见不良反应包括胃肠反应、金属味、头痛、罕见神经毒性(周围神经病变、共济失调)等。与酒精合用可发生双硫仑样反应;与华法林等抗凝药物可能相互作用,需监测凝血功能。 |
抗病毒药
抗疱疹病毒药
| 类别 | 要点 |
|---|---|
| 药物 | 阿昔洛韦(Acyclovir):核苷类(鸟嘌呤)抗病毒药。 |
| 作用机制 | 1) 依赖病毒胸苷激酶首磷酸化,随后由细胞激酶转为二、三磷酸。 2) 阿昔洛韦三磷酸竞争性抑制病毒DNA聚合酶并被掺入到新合成的DNA链中,导致链终止,从而抑制病毒复制。 |
| 抗病毒谱 | 对单纯疱疹病毒(HSV‑1、HSV‑2)和水痘‑带状疱疹病毒(VZV)有良好活性。对巨细胞病毒(CMV)活性有限,常用更活跃的药物(如更昔洛韦/更昔洛韦衍生物)治疗CMV。 |
| 临床应用 | 首选用于HSV和VZV感染的治疗与预防:外用/口服/静脉制剂用于皮肤科、眼科(单纯疱疹性角膜炎)、带状疱疹、严重系统感染及免疫受损者的预防。对HIV或慢性乙型肝炎并非常规治疗药物(不能作为HIV或HBV的抗病毒主药)。 |
| 给药与注意事项 | 主要经肾脏清除,肾功能不全需调整剂量;静脉用药可导致晶体肾病和肾功能恶化(需充分补液、缓慢滴注)。常见不良反应:头痛、恶心、肾功能异常、静脉炎;罕见中枢神经系统症状(谵妄、幻觉)在老年或肾功能差者更常见。 |
| 耐药与替代 | 耐药常因病毒胸苷激酶突变或DNA聚合酶改变。对阿昔洛韦耐药的HSV/VZV可选用法匹拉韦(famciclovir)、伐昔洛韦(valacyclovir)或静脉用更活跃药物(如泛昔洛韦、福昔洛韦或更昔洛韦等,视病原和耐药机制而定)。 |
抗流感病毒药
| 项目 | 要点 |
|---|---|
| 代表药物 | 金刚烷胺(amantadine) 金刚乙胺(rimantadine) |
| 抗病毒谱 | 主要对A 型流感病毒有特异性活性;大剂量时可抑制部分 B 型流感或其他病毒的体外活性(临床意义有限)。 |
| 作用机制 | 抑制病毒 M2 离子通道(阻断质子流入),阻止病毒脱壳与核酸释入宿主细胞胞质,主要在病毒复制的早期发挥作用,从而阻断 A 型流感病毒入侵-脱壳过程。 |
| 临床应用 | 用于流行性感冒(主要为 A 型)的治疗与暴露后预防。但需注意:全球范围内 A 型流感对这类药物的耐药率已显著上升,限制其临床使用,多数指南不再优先推荐。 |
| 药代与不良反应(要点) | 金刚乙胺代谢较快、系统副作用较少;金刚烷胺易通过血脑屏障,中枢神经系统副作用(头晕、失眠、幻觉)更常见;老年人、肾功能不全需调整剂量并谨慎使用。 |
抗肝炎病毒药
| 项目 | 内容 |
|---|---|
| 代表药物 | 拉米夫定(Lamivudine, 3TC) 核苷类逆转录酶抑制剂(胞嘧啶类似物),口服制剂,常见片剂剂量:100 mg/片。 |
| 作用机制 | 经细胞内磷酸化为活性三磷酸形式,竞争性抑制HBV DNA聚合酶(含反转录酶活性),并通过链终止阻断HBV前基因组RNA逆转录为DNA,从而抑制病毒复制。 |
| 主要临床应用 | 慢性乙型肝炎(CHB)抗病毒治疗:用于需抗病毒治疗的HBV感染者以降低HBV DNA、改善肝功能和减轻肝纤维化进展。亦用于对某些患者的阻断性或替代性治疗(如在资源受限地区或与其他药物合用)。 |
| 给药与剂量(成人) | 常规剂量:100 mg qd(每日一次)。对肾功能不全需调整剂量或延长给药间隔;终末期肾病/血液透析患者需按CrCl调整并于透析后补给。 |
| 耐药问题 | 高频耐药突变:YMDD位点突变(M204V/I ± L180M),长期单药治疗耐药率较高,临床上常需更换或加用高屏障药物(如替诺福韦、恩替卡韦)。 |
| 不良反应 | 常见:头痛、疲乏、恶心;少见但严重:乳酸性酸中毒/重度肝脏脂肪变性(罕见)、胰腺炎(极少)。停药可出现HBV反弹/肝功能加重(需密切随访)。 |
| 监测与注意事项 |
|
| 孕妇与哺乳 | 妊娠中常用以降低母婴传播风险的抗病毒药物之一;在必要时可使用(权衡利弊并遵循指南)。哺乳期可分泌入乳汁,临床使用需评估风险与益处。 |
| 与其他药物的临床整合 | 因耐药问题,临床常与高屏障核苷/核苷酸类药物(如替诺福韦/恩替卡韦)联合或用后者取代。对已发生YMDD突变者,优先选择对该突变仍有效的药物策略。 |
抗真菌药
抗生素类药
| 类别 | 要点 |
|---|---|
| 药物 | 两性霉素 B(Amphotericin B) 天然多烯抗真菌药 |
| 作用机制 | 与真菌细胞膜中的麦角固醇结合,形成孔道或改变膜完整性,增加膜通透性导致细胞内容物外泄,具有杀菌/抑菌作用。 |
| 临床应用 | 常以静脉滴注给药,适用于全身性或深部真菌感染(如侵袭性曲霉病、念珠菌血症、隐球菌脑膜炎等)。 |
唑类
| 项目 | 要点与说明 |
|---|---|
| 代表药物 | 氟康唑(Fluconazole) — 口服/静脉用广谱三唑类抗真菌药,药代动力学稳定,能穿透中枢神经系统。 |
| 作用机制 | 抑制真菌14α-脱甲基酶(CYP450),干扰麦角固醇生物合成→导致膜结构破坏、膜通透性增加,抑制真菌生长或致死。 |
| 临床应用 | 对隐球菌属、念珠菌属、球孢子菌属等有效;为治疗艾滋病相关隐球菌性脑膜炎的首选药,与氟胞嘧啶合用可增强疗效。亦用于口腔/食管/生殖道念珠菌病、系统性念珠菌感染及免疫受损患者的预防。注意与其他CYP底物的药物相互作用并监测肝功能。 |
抗结核药、抗疟药与抗恶性肿瘤药
药物分类及药理作用。
考题
- 【例1】可引起周围神经炎的药物是 A. 利福平 B. 异烟肼 C. 阿昔洛韦 D. 吡嗪酰胺 E. 卡那霉素
- 【例2】能诱发“流感样综合征”的药物是 A. 利福平 B. 多粘菌素 C. 链霉素 D. 庆大霉素 E. 头孢孟多
- 【例3】治疗结核病选用的药物是 A. 磺胺嘧啶 B. 四环素 C. 异烟肼 D. 甲氧苄啶 E. 环丙沙星
- 【例4】治疗铜绿假单胞菌感染首选的药物是 A. 磺胺嘧啶 B. 四环素 C. 异烟肼 D. 甲氧苄啶 E. 环丙沙星
- 【例5】治疗嗜肺军团菌肺炎选用的药物是 A. 庆大霉素 B. 乙胺嘧啶 C. 头孢噻肟 D. 利福平 E. 红霉素
- 【例6】用于治疗结核病和麻风病的药物是 A. 庆大霉素 B. 乙胺嘧啶 C. 头孢噻肟 D. 利福平 E. 红霉素
- ✨【例7】预防性抗疟疾药是 A. 乙胺嘧啶 B. 乙胺丁醇 C. 异烟肼 D. 氯喹 E. 伯氨喹
- 【例8】控制普通型疟疾发作多选用的药物是 A. 乙胺嘧啶 B. 氯喹 C. 奎宁 D. 哌喹 E. 伯氨喹
- 【例9】防止疟疾复发选用的药物是 A. 乙胺嘧啶 B. 氯喹 C. 奎宁 D. 哌喹 E. 伯氨喹
- 【例10】控制间日疟发作的首选药物是 A. 吡喹酮 B. 乙胺嘧啶 C. 氯喹 D. 伯氨喹 E. 奎宁
- 【例11】防止疟疾复发的药物是 A. 吡喹酮 B. 乙胺嘧啶 C. 氯喹 D. 伯氨喹 E. 奎宁
- 【例12】阿霉素 A. 对心脏的影响最大 B. 对肾的影响最大 C. 对肺毒性最大 D. 对骨髓抑制最重 E. 发生腹泻最重
- ✨【例13】顺铂 A. 对心脏的影响最大 B. 对肾的影响最大 C. 对肺毒性最大 D. 对骨髓抑制最重 E. 发生腹泻最重
- 【例14】常引起周围神经炎的化疗药是 A. 环磷酰胺 B. 甲氨蝶呤 C. 长春新碱 D. 阿霉素 E. L-门冬酰胺酶
- 【例15】常引起心脏毒性的化疗药是 A. 环磷酰胺 B. 甲氨蝶呤 C. 长春新碱 D. 阿霉素 E. L-门冬酰胺酶
基础
抗结核药
异烟肼
| 区域 | 要点 | 详细描述 |
|---|---|---|
| 药理作用 | 对结核分枝杆菌的选择性杀菌作用 | 主要杀灭生长旺盛的活动期结核分枝杆菌,抑制细菌细胞壁合成相关代谢(为一线杀菌药)。 |
| 临床应用 | 一线抗结核药;联合用药为常规原则 | 适用于各种类型结核病。早期轻症或预防时可单药短期使用;规范化治疗必须与其他抗结核药联合应用,以防或延缓耐药产生。 |
| 不良反应 | 神经系统 | 周围神经炎为最常见,表现为手足麻木、感觉异常、肌肉震颤、步态不稳;大剂量可引起头痛、头晕、兴奋、视神经炎,严重可致中毒性脑病或精神病。 预防:常规补充维生素B6(吡哆醇)。 |
| 肝脏毒性 | 可引起肝损伤,用药期间应定期监测肝功能;已有肝功能不良者慎用或调整方案。 | |
| 其他 | 可见皮疹、发热、胃肠道反应、粒细胞减少、血小板减少、溶血性贫血、脉管炎、关节炎样综合征等。遇严重不良反应停药并处理。 |
利福平
| 区域 | 要点 | 说明 |
|---|---|---|
| 药理作用 | 广谱且强效的抗菌作用 | 对静止期和繁殖期细菌均有效;能增强链霉素、异烟肼的抗菌活性;主要通过抑制细菌RNA聚合酶发挥杀菌作用(肠杆菌科、革兰阳性及部分分枝杆菌敏感)。 |
| 临床应用 | 结核病 | 与其他抗结核药物联合使用:初治者常与异烟肼联合;复发或复杂病例与乙胺丁醇、吡嗪酰胺等联合治疗。 |
| 麻风病与耐药细菌感染 | 用于麻风病及对利福平敏感的细菌感染(包括部分耐药金黄色葡萄球菌)。 | |
| 胆道感染 | 因在胆汁中浓度高,可用于重症或难治性胆道感染作为辅助药物。 | |
| 眼科局部用药 | 可用于沙眼、急性结膜炎和部分病毒性角膜炎的局部治疗(外用制剂)。 | |
| 不良反应 | 胃肠道反应(常见) | 恶心、呕吐、腹痛、腹泻,通常轻度且可耐受。 |
| 肝脏毒性(需警惕) | 长期或大剂量使用可致肝损伤:黄疸、肝肿大、肝功能减低等,使用期间应监测肝功能。 | |
| 类流感样反应(间断大剂量) | 大剂量间隔使用可诱发发热、寒战、头痛、肌肉痛等类似感冒的症状。 |
乙胺丁醇
| 区域 | 要点 | 说明 |
|---|---|---|
| 药理作用 | 抑菌靶点 | 与 Mg2+ 络合,阻止亚精胺(spermidine)与 Mg2+ 结合,从而干扰细菌 RNA 合成,抑制繁殖期结核分枝杆菌生长。 |
| 选择性 | 主要对结核分枝杆菌有效,对多数非结核细菌无效。 | |
| 耐药与联合用药 | 单药耐药风险 | 单独使用易产生耐药,因而临床常与其他抗结核药联合使用;无明显交叉耐药(与其他常用抗结核药间)。 |
| 常用联合方案 | 用于初治肺结核常与异烟肼合用;复治患者常与利福平和链霉素等联合。(根据具体指南调整用药方案) | |
| 临床适应证 | 适应证 | 各型肺结核及肺外结核;对经链霉素和异烟肼治疗无效或耐药的患者尤为重要。 |
吡嗪酰胺
| 区域 | 项目 | 内容 |
|---|---|---|
| 药理作用 | 抗结核活性 | 在酸性环境中对结核分枝杆菌具有强烈抑制及杀灭作用(对静止期或巨噬细胞内菌株活性佳) |
| 临床应用 | 用途 | 用于结核病化学治疗方案中,单用易致耐药,常与异烟肼、利福平等联合使用以缩短疗程并防止耐药 |
| 注意事项 | 耐药与联合用药 | 单药治疗禁用;出现耐药时需依药敏调整方案;注意肝功能监测与潜在副作用(如高尿酸血症、肝损伤) |
抗疟药
青氯减症,伯伯休眠
- 疟原虫生活史及各类抗疟药的作用部位。裂殖体破坏红细胞,释放裂殖子、疟色素及其他代谢产物,刺激机体引起寒战、高热等症状,即疟疾发作。
- 常考抗疟药的比较
| 项目 | 氯喹 | 青蒿素 | 伯氨喹 | 乙胺嘧啶 |
|---|---|---|---|---|
| 属于 | 控制症状的药物 | 控制症状的药物 | 控制复发和传播的药物 | 病因性预防的药物 |
| 主要杀灭 | 红细胞内期裂殖体,对多种疟原虫有效 | 红细胞内期裂殖体,起效快、杀灭迅速 | 肝脏休眠子(间日、卵形疟)和配子体 | 抑制疟原虫的增殖(阻断蚊内发育/传播) |
| 无效 | 对孢子子、休眠子及配子体无效 | 对红细胞外期寄生体无效 | 对红细胞内期裂殖体无效 | 对已成熟的裂殖体无效(对配子体间接作用) |
| 临床应用 | 迅速有效控制疟疾临床发作,非首选病因性预防,可用于短期控制,亦用于减缓传播 | 迅速有效控制临床发作,用于氯喹/多药耐药的恶性疟;可透过血脑屏障,用于脑性疟救治 | 防止远期复发(根治良性疟),与红细胞期抗疟药合用;可阻断传播 | 主要用于病因性预防/阻断传播,不能直接杀灭配子体但阻止蚊体内发育 |
| 特点 | 起效快、疗效高;24–48小时内症状消失,48–72小时血中裂殖体减少/消失 | 起效非常快,48小时内血中疟原虫显著减少或消失 | 耐药株少见;用于根治与阻断传播 | 起效相对缓慢,常用于病因性预防/阻断传播 |
| 不良反应 | 罕见 | 罕见 | 高铁血红蛋白血症、急性溶血(G6PD缺乏时风险增高) | 巨幼细胞性贫血、粒细胞减少 |
青蒿素
| 区域 | 要点 | 详细说明 |
|---|---|---|
| 药理/机制 | 靶向期 | 选择性杀灭红细胞内裂殖体(intraerythrocytic schizonts),对红细胞外游离期疟原虫无效。 |
| 作用机制(可能) | 过氧化架构在血红素或Fe2+催化下产生自由基,自由基损伤疟原虫膜与线粒体等细胞结构,导致快速死亡。 | |
| 临床应用 | 适应证与优势 | 首选或备用治疗耐氯喹/多药耐药恶性疟(包括重症脑性疟);能穿过血脑屏障,对脑性疟疗效较好,起效快、清除寄生虫速度高。 |
氯喹
| 区域 | 要点 | 详细说明 |
|---|---|---|
| 药理作用 | 抗疟作用 | 对红细胞内裂殖体具强力杀灭作用,浓集于被疟原虫侵入的红细胞内,起效快、疗效高;对孢子、休眠体和配子体无效,不能阻断远期复发或传播。 |
| 抗肠道外阿米巴作用 | 能杀灭阿米巴滋养体,用于肠道外阿米巴感染的药理基础。 | |
| 免疫抑制作用 | 大剂量可抑制免疫反应,在类风湿关节炎、系统性红斑狼疮等疾病中有时作为免疫调节剂使用(剂量与适应证有限)。 | |
| 临床应用 | 主要适应证:疟疾(红细胞期) | 对各种疟原虫的红细胞内期裂殖体有效,能迅速控制临床症状;不用于病因性预防远期复发或阻断传播(因对孢子/休眠体无效)。 |
伯氨喹
| 区域 | 要点 | 详细说明 |
|---|---|---|
| 药理作用 | 杀灭肝内休眠虫 | 对间日疟(P. vivax)和卵形疟(P. ovale)肝脏休眠子(hypnozoite)有较强杀灭作用,可预防复发;对红细胞期无直接疗效。 |
| 药理作用 | 杀灭配子体,阻断传播 | 能杀灭各种疟原虫的配子体(gametocytes),从而阻止疟疾通过蚊媒传播。 |
| 临床应用 | 预防和根治复发 | 是防治疟疾远期复发的主要药物;常与对红细胞内期有效的抗疟药合用以根治良性疟并减少耐药发生。 |
| 临床应用 | 联合用药策略 | 应与红细胞内期抗疟药合用(例如氯喹或喹啉类对敏感株)以同时清除肝内休眠子和血液期寄生虫;单用对血浆期无效。 |
乙胺嘧啶
| 区域 | 要点 | 说明 |
|---|---|---|
| 药理作用 | 抑制二氢叶酸还原酶(DHFR) | 阻止二氢叶酸转变为四氢叶酸,抑制核酸合成,对疟原虫DHFR的亲和力远高于人体同酶,因而选择性抑制疟原虫增殖。 对已发育成熟的裂殖体无效,临床症状缓解起效较慢。 |
| 临床应用 | 适应证 | 常用于疟疾的病因性预防(作为抗疟策略的一部分)。 |
抗恶性肿瘤药
- 细胞毒药物
| 常用药物 | 作用机制 | 临床应用 / 主要不良反应 |
|---|---|---|
| 甲氨蝶呤 (methotrexate) | 抑制二氢叶酸还原酶 | 临床:急性白血病、鳞状上皮癌;鞘内注射治疗中枢神经系统白血病 不良反应:消化道反应、骨髓抑制最突出、肝肾损害 |
| 氟尿嘧啶 (5‑FU) | 抑制胸苷酸合成酶(阻断胸苷酸合成) | 临床:消化道系统肿瘤、乳腺癌、宫颈癌、卵巢癌、鳞癌、膀胱癌、头颈部肿瘤 不良反应:骨髓抑制、消化道毒性较大 |
| 6‑巯基嘌呤 (6‑MP) | 抑制嘌呤核苷酸代谢/互变 | 临床:急性淋巴细胞白血病、淋巴瘤等 不良反应:骨髓抑制、消化道黏膜损害 |
| 羟基脲 (hydroxyurea) | 抑制核苷酸还原酶 | 临床:慢性粒细胞白血病、黑色素瘤等 不良反应:骨髓抑制、消化道反应、致畸 |
| 环磷酰胺 (cyclophosphamide) | 烷化剂——导致DNA交联 | 临床:恶性淋巴瘤、多发性骨髓瘤、急性淋巴细胞白血病、肺癌、乳腺癌、卵巢癌、神经母细胞瘤、移植抑制等 不良反应:骨髓抑制、恶心呕吐、脱发、出血性膀胱炎 |
| 阿霉素 (doxorubicin) | 嵌入DNA并抑制拓扑异构酶Ⅱ(破坏DNA复制/转录) | 临床:急性白血病、淋巴瘤、乳腺癌、肺癌等 不良反应:心脏毒性(累及性心肌病)、骨髓抑制 |
- 靶向药物
| 药物 | 要点 | 详情 |
|---|---|---|
| 利妥昔单抗 (Rituximab) | 单克隆抗体 介导细胞毒性 | 作用机制:抗CD20单抗,主要通过ADCC、CDC和直接凋亡清除B细胞。 靶点:CD20(B细胞分化抗原)。 临床应用:弥漫性/滤泡性等非霍奇金淋巴瘤、慢性淋巴细胞白血病等。 主要不良反应:输注相关反应(发热、畏寒、寒战、头痛)、感染风险、罕见心血管/皮肤反应。 |
| 曲妥珠单抗 (Trastuzumab) | 靶向HER2的单抗 抑制受体信号 | 作用机制:结合HER2外部结构域,阻断受体介导的信号转导并介导ADCC,抑制肿瘤细胞增殖。 靶点:HER2/ERBB2(表皮生长因子受体家族成员)。 临床应用:HER2阳性转移性或早期乳腺癌,部分HER2阳性胃癌。 主要不良反应:输注反应(发热、寒战、恶心头痛)、心功能受损(心衰风险),少见肺毒性。 |
| 伊马替尼 (Imatinib) | 小分子酪氨酸激酶抑制剂 靶向融合/突变激酶 | 作用机制:竞争性抑制ATP结合位点,抑制BCR‑ABL、c‑KIT、PDGFR等的酪氨酸激酶活性,阻断肿瘤细胞信号传导与增殖。 靶点:BCR‑ABL、c‑KIT、PDGFR。 临床应用:慢性粒细胞白血病(CML)、胃肠道间质瘤(GIST)等。 主要不良反应:消化道反应(恶心、腹泻)、液体潴留、骨髓抑制、肝功能异常、罕见心肌毒性。 |
- 免疫治疗药物
| 药物 | 作用机制 / 靶点 | 临床适应证 / 主要不良反应 |
|---|---|---|
| 伊匹木单抗 (ipilimumab) | CTLA‑4 抑制 增强早期T细胞活化 | 适应证:转移性黑色素瘤 主要不良反应:疲乏、腹泻、瘙痒、皮疹(免疫相关毒性以肠炎、皮炎、内分泌炎症为主) |
| 尼伏单抗 (nivolumab) | 程序性死亡受体‑1(PD‑1)拮抗 恢复耗竭T细胞抗肿瘤活性 | 适应证:黑色素瘤、非小细胞肺癌等(多种实体瘤) 主要不良反应:皮疹、肺炎、肝炎、肾炎(免疫相关不良反应:肺、肝、肾、内分泌等) |
| 派姆单抗 (pembrolizumab) | 人源化PD‑1 单抗(抗PD‑1) 恢复T细胞功能,广泛适应证 | 适应证:转移性黑色素瘤等多种实体瘤 主要不良反应:疲劳、咳嗽、恶心、瘙痒、皮疹(同类免疫相关不良反应) |
| 阿替利珠单抗 (atezolizumab) | PD‑L1 抑制(抗PD‑L1) 阻断肿瘤细胞/免疫细胞上的PD‑L1与PD‑1相互作用 | 适应证:转移性尿路上皮癌等 主要不良反应:疲劳、食欲减退、尿路感染、发热(并可见肺炎、肝炎等免疫相关毒性) |
| 度伐利尤单抗 (durvalumab) | PD‑L1 抑制(抗PD‑L1) 解除PD‑L1介导的免疫抑制 | 适应证:转移性尿路上皮癌等 主要不良反应:疲劳、咳嗽、恶心(并可出现免疫相关肺/肝/内分泌不良反应) |