Last updated: 22 Aug 25 10:08:00 (Asia/Shanghai)
血液系统疾病
This tutorial is powered by Bensz/黄伟斌
速记
| 主题 | 特征性表现/关键鉴别 | 首选检查与治疗要点 |
|---|---|---|
| 贫血分型 | 小细胞低色素(缺铁、地中海);大细胞(巨幼、MDS);正细胞(急失血、溶血) | 红细胞指数(MCV/MCH/MCHC)、网织红细胞、铁代谢;缺铁首选口服二价铁,B12缺乏先补维生素B12 |
| 白细胞减少 | 多由病毒/药物/骨髓病引起,感染风险与ANC密切相关 | 停可疑药、病因学筛查;高危/发热经验性抗菌+G‑CSF |
| 粒细胞缺乏症 | 高热、口腔坏死性溃疡,易败血症;常见于药物所致 | 立即广谱抗生素、G‑CSF、隔离;必要时粒细胞输注 |
| MDS | 无效造血、三系减少,MCV常大;易“转白” | 骨髓形态+细胞遗传/分子;低危支持/来那度胺(5q‑),中高危阿扎胞苷/地西他滨;根治异基因移植 |
| 急性白血病 | 贫血+出血+感染,肝脾淋巴结肿大;APL易DIC | 骨髓原始细胞≥20%+免疫分型;AML用DA诱导,APL用全反式维甲酸±砷剂,ALL行中枢预防 |
| 淋巴瘤 | HL有Reed‑Sternberg细胞、连续性播散;NHL多中心非连续 | 切除活检+免疫分型;HL首选ABVD±放疗;DLBCL首选R‑CHOP |
| 多发性骨髓瘤 | CRAB:高钙、肾损、贫血、骨痛/溶骨 | 蛋白电泳/免疫固定/游离轻链+骨髓;一线硼替佐米+来那度胺+地塞米松,适合者自体移植,骨病双膦酸盐 |
| 出血性疾病 | 皮肤黏膜出血(血小板)vs 关节肌肉出血(因子);DIC混合型 | ITP首选激素;血友病因子替代;vWF病去氨加压素;DIC病因治疗+成分纠正 |
| 输血 | 成分输血优先,严格三查八对 | RBC:Hb<70 g/L或有缺氧证据;PLT:按阈值与手术;FFP/冷沉淀:凝血障碍;急性溶血:立停、复核、补液利尿、纠正DIC |
- 白细胞减少是指成人外周血白细胞总数持续低于 4.0x109/L。中性粒细胞减少症是指成人外周血中性粒细胞绝对计数低于2.0×109/L。粒细胞缺乏症是指外周血中性粒细胞绝对计数低于0.5x109/L。
- 不同类型的贫血的鉴别要点
| 病因/类型 | 典型实验室/形态学提示 | 鉴别要点(临床+辅助检查) |
|---|---|---|
| 缺铁性贫血 | 小细胞低色素(MCV↓) RDW↑,网织红细胞正常或↓ | 病因提示:慢性失血(消化道、妇科)或摄入不足。血清铁↓,TIBC↑,血清铁蛋白↓(敏感特征)。外周血涂片见小红细胞与椭圆形红细胞;反应性骨髓铁染色(Prussian blue)缺铁。对比:与炎症性贫血的铁蛋白相反(炎症时铁蛋白↑)。 |
| 慢性疾病/炎症性贫血(ACD) | 常为小至正细胞 铁↓或正常;TIBC↓;血清铁蛋白正常或↑ | 病史提示慢性炎症、感染、肿瘤或自身免疫病。促铁调素↑导致铁被滞留于网状内皮系;网织红细胞正常或↓。与缺铁性贫血鉴别用铁蛋白、TIBC和炎症标志物(CRP/ESR)。若不明确可行骨髓铁染色或治疗性铁剂试验(谨慎)。 |
| 巨幼细胞性贫血(叶酸/B12缺乏) | 巨红细胞性/MCV↑ 周边白细胞或血小板可↓,巨核细胞/高RDW | 典型表现:舌炎、神经症状(B12缺乏可有周围神经病变/脊髓后索受累)。血清维生素B12↓或叶酸↓;同型半胱氨酸↑(对B12和叶酸均升高),甲基丙二酸↑提示B12缺乏。鉴别药物(甲氨蝶呤、抗癫痫药)、肠吸收障碍(吸收不良/手术史)或恶性病变。 |
| 溶血性贫血(遗传/获得) | 溶血指标阳性:间接胆红素↑、LDH↑、游离血红蛋白↑、血红蛋白尿/尿胆原↑、网织红细胞↑ | 外周涂片可见球形红细胞、碎裂细胞、靶形细胞或胞浆内包涵体。直接抗人球蛋白试验(Coombs)阳性提示获得性AIHA;G6PD缺乏表现为发作性溶血、伴包涵体和Heinz小体;遗传性球形细胞症提示家族史、红细胞浸透压试验异常。关键是区分所得性与先天性并寻找触发因素(药物、感染、温度变化)。 |
| 地中海/珠蛋白生成障碍(地中海贫血) | 小细胞低色素,但网织红细胞不高或相对正常 Hb电泳或基因检测阳性 | 家族史、青年起病、脾肿大常见。外周血见靶形细胞、微小红细胞;铁染色可显示骨髓铁正常或增多(需防误诊为缺铁)。诊断依靠血红蛋白电泳(成人)或分子诊断。输血依赖者有铁负荷表现。 |
| 再生障碍性贫血/骨髓衰竭 | 全细胞减少或孤立性细胞系减少;网织红细胞↓ | 临床:感染、出血倾向、无明显溶血或营养缺陷证据。骨髓象呈低细胞或脂肪髓,需排除药物/毒物、病毒(如EBV、HBV、HCV、HIV)、自身免疫及遗传性综合征。骨髓活检是确诊金标准。 |
| 慢性肾性贫血 | 通常为轻-中度正细胞性贫血;网织红细胞低或正常 | 肾功能异常史、肌酐↑;EPO水平低。对比其他原因,纠正肾功能或给予促红素(ESA)可改善;并注意铁状态以保证ESA疗效。 |
| 环状或铁粒幼细胞性贫血(造血合成障碍/铅中毒) | MCV可变;骨髓有环状铁粒幼细胞 | 考虑药物、酒精、铅暴露或先天代谢缺陷。血铅升高提示铅中毒;骨髓铁染色显示过多铁与环状幼红细胞提示铁粒幼细胞性贫血或MDS。鉴别需骨髓检查与暴露史。 |
| 缺乏/营养相关(混合原因) | 可表现为小或大细胞,RDW↑ | 常见混合:缺铁合并叶酸/B12缺乏或慢性病合并营养不良。通过全面营养指标(铁、铁蛋白、B12、叶酸)与临床史(饮食、吸收、慢性病)综合判断。 |
| 药物/毒物导致贫血 | 表现多样(溶血/再生障碍/巨幼细胞等) | 重要提示:详细用药史(抗肿瘤药、抗菌、免疫抑制剂、抗癫痫药、化学物质)。时间关联性强,停药后观察或行靶向检查(Coombs、骨髓)以确定机制。 |
- HL vs. NHL
| 项目 | 霍奇金淋巴瘤(HL) | 非霍奇金淋巴瘤(NHL) |
|---|---|---|
| 起源与病理学 | 由肿瘤性朗格汉斯细胞/Reed–Sternberg细胞相关,病理特征为含有典型的Reed–Sternberg细胞与丰富反应性细胞浸润,亚型分为结节硬化型、混合细胞型等。 | 源自不同成熟阶段的B细胞或T/NK细胞,病理类型多样(弥漫大B细胞淋巴瘤、滤泡性淋巴瘤、T细胞淋巴瘤等),无Reed–Sternberg细胞,免疫表型和分子特征差异大。 |
| 临床特点 | 典型表现为无痛性淋巴结肿大(常为颈部或锁骨上),常伴发热、盗汗、体重下降(B症状);多见于青壮年,有时出现瘙痒或酒精诱发淋巴结痛。 | 表现多样:从局限的可触及肿块到广泛结外侵犯(胃肠道、皮肤、脾、骨髓等);B症状亦可出现,但年龄分布更宽(儿童到老年均可),进展与侵袭性相关。 |
| 影像学与分布 | 常见纵隔/纵隔前肿块(特别是结节硬化型),病程早期沿淋巴结连锁顺序扩展,骨髓早期受累较少。 | 分布不规则,常可见多发结外病灶与弥漫性结外受累;淋巴结间可不按固定途径扩展,骨髓受累概率随亚型可高。 |
| 免疫组化/分子 | RS细胞通常CD15+/CD30+,PAX5弱阳性;B/T细胞标记常呈不典型表达,EBV关联在某些亚型中常见。 | 依亚型不同而异:多数B细胞NHL表达CD19/CD20/CD79a等,T细胞NHL表达CD3等;分子检测(BCL2/BCL6/MYC等)和基因重排对诊断与预后重要。 |
| 分期与预后 | 按Ann Arbor分期;多数早期患者对放/化疗敏感,预后良好(尤其限局期)。 | 异质性大:低度恶性(如滤泡性)可为慢性演变,高度恶性(如弥漫大B细胞)需积极化疗,预后依病理亚型与分期显著不同。 |
| 治疗原则 | 以放疗+化疗为主(如ABVD方案),部分早期可单独放疗或限期化疗;难治/复发者可行自体造血干细胞移植或免疫治疗(PD-1抑制剂在某些亚型有效)。 | 以化疗±靶向治疗为主(例如R-CHOP对CD20阳性B细胞NHL),部分局限病变可行放疗;对不同分期/亚型个体化方案及靶向/免疫治疗广泛应用。 |
| 诊断要点 | 淋巴结活检找到典型Reed–Sternberg细胞并做免疫组化(CD15+/CD30+)是确诊关键。 | 需组织学分型、免疫表型(CD20/CD3等)、基因重排/分子检测以明确亚型;无RS细胞,诊断依赖病理学与免疫学证据。 |
- 血液系统恶性肿瘤常用化疗药物
| 药物 | 英文 | 药理作用 | 临床应用 | 常见方案 |
|---|---|---|---|---|
| 阿糖胞苷 | Cytarabine / Ara‑C | 为核苷类似物,经磷酸化后被掺入DNA,抑制DNA聚合酶并导致链终止;细胞周期S期特异。 | 急性髓系白血病(AML)的一线诱导与巩固治疗;也用于某些淋巴瘤或中枢神经系统浸润(鞘内给药)。 | 3+7方案(高剂量或标准剂量Ara‑C联合蒽环类);高剂量Ara‑C巩固;鞘内注射剂量用于CNS预防/治疗。 |
| 蒽环类(柔红霉素/阿霉素/伊达比星) | Daunorubicin / DNR;Doxorubicin;Idarubicin | 与DNA插入并抑制拓扑异构酶Ⅱ,产生活性氧导致DNA断裂;非周期特异。 | AML、ALL、淋巴瘤及多种实体瘤的诱导或联合方案。 | 与Ara‑C组成AML诱导方案(如DNR+Ara‑C);急性白血病或淋巴瘤常用组合(如CHOP中用Doxorubicin)。 |
| 环磷酰胺 | Cyclophosphamide / CPA | 烷化剂,经肝代谢激活,导致DNA交联与断裂;非周期特异,免疫抑制效应显著。 | 淋巴瘤、白血病、骨髓移植前方案及免疫相关血液病。 | CHOP(与多柔比星、长春新碱、泼尼松)用于非霍奇金淋巴瘤;高剂量用于造血干细胞移植预处理。 |
| 长春新碱 | Vincristine / VCR | 抑制微管形成,阻断有丝分裂的M期;神经毒性(周围神经病)为主要不良反应。 | ALL、非霍奇金淋巴瘤、某些急性白血病方案中的关键药。 | 常见于CHOP、Hyper‑CVAD等方案;注意伴随苯妥英等需调整剂量。 |
| 泼尼松 / 泼尼松龙 | Prednisone / Prednisolone | 糖皮质激素:诱导淋巴细胞凋亡、消炎免疫抑制;对淋巴系肿瘤有直接杀伤作用。 | 淋巴瘤、ALL、某些多发性骨髓瘤方案的重要组成部分;用于肿瘤相关症状缓解。 | CHOP、CVP、Hyper‑CVAD等方案中的组成药;也用作短期减瘤或支持治疗。 |
| 利妥昔单抗 | Rituximab / RTX | 抗CD20单抗,通过抗体依赖性细胞介导细胞毒(ADCC)、补体依赖性细胞溶解(CDC)及直接凋亡诱导起效。 | CD20阳性B细胞淋巴瘤、慢性淋巴细胞白血病(部分病例)。 | R‑CHOP(与CHOP联合)为弥漫性大B细胞淋巴瘤首选;维持治疗或与化疗联合用于FL等。 |
| 伊马替尼 | Imatinib / TKI | 小分子酪氨酸激酶抑制剂,抑制BCR‑ABL、c‑KIT、PDGFR等;为靶向治疗。 | 慢性髓性白血病(CML,BCR‑ABL阳性)、某些胃肠间质瘤(GIST)。 | 口服维持疗法(CML一线),剂量依病情(如400 mg/d常见);与其他TKI比较有替代选择。 |
| 氟达拉滨 | Fludarabine / F‑araA | 腺苷类似物,抑制DNA聚合、诱导断裂,主要用于淋巴细胞系列肿瘤;有显著免疫抑制。 | 慢性淋巴细胞白血病(CLL)、某些低级别淋巴瘤。 | 常与环磷酰胺、利妥昔单抗联合(FCR方案)用于适合的CLL患者。 |
| 甲氨蝶呤 | Methotrexate / MTX | 叶酸拮抗剂,抑制二氢叶酸还原酶,阻断嘧啶/嘌呤合成;对增殖快的细胞敏感。高剂量具穿透CNS能力需碱化尿并监测药物浓度。 | 白血病(尤其ALL)高剂量系统治疗或鞘内用于CNS防治;也用于恶性淋巴瘤、骨肉瘤等。 | 高剂量MTX联合亚叶酸拯救用于ALL或某些淋巴瘤的CNS方案;低剂量作为维持/联合方案。 |
| 阿扎胞苷 / 地西他滨 | Azacitidine / Decitabine | 核苷类似物,抑制DNA甲基转移酶(DNMT),导致去甲基化和基因再表达,兼具细胞毒性作用。 | 骨髓增生异常综合征(MDS)、某些老年AML作为低强度治疗。 | Azacitidine 7天循环皮下或静脉给药;Decitabine常规5天方案或低剂量方案用于不适合强化化疗者。 |
| L‑天冬酰胺酶 | L‑Asparaginase | 分解天冬氨酸,致使肿瘤细胞因缺乏合成能力而死亡;对某些淋巴瘤/ALL高度敏感。 | 急性淋巴细胞白血病(特别是童年ALL)和部分淋巴瘤。 | 常见于儿童ALL方案(如ALL‑BFM、Pediatric protocols)及成人改良方案;注意过敏和胰腺炎、凝血异常。 |
| 依托泊苷 | Etoposide / VP‑16 | 拓扑异构酶Ⅱ抑制剂,诱导DNA双链断裂,细胞周期对S/G2期敏感。 | 某些淋巴瘤、急性白血病、霍奇金淋巴瘤复发方案及造血干细胞移植前方案。 | 与顺铂或铂类、烷化剂联合用于复发/难治性淋巴瘤或小细胞癌相关方案;口服/静脉制剂均可。 |
| 硼替佐米 / 胞浆蛋白酶体抑制剂 | Bortezomib / BZM | 可逆性蛋白酶体抑制剂,阻断NF‑κB信号与蛋白降解,诱导肿瘤细胞凋亡。 | 多发性骨髓瘤一线与复发治疗的核心药物。 | Bortezomib‑含方案(如VRd:Bortezomib+Lenalidomide+Dexamethasone);皮下或静脉给药,注意带状疱疹预防及周围神经病变管理。 |
基础
贫血
病因
- 生成不足性:铁缺乏(最常见)、维生素B12/叶酸缺乏(巨幼细胞性)、慢性病性贫血、慢性肾病(促红素减少)、骨髓造血抑制或受侵(再生障碍性贫血、骨髓增生异常性肿瘤/综合征、肿瘤浸润)、内分泌障碍等。
- 溶血性:红细胞内在缺陷(膜病、酶缺陷如G6PD、血红蛋白病如地中海贫血/镰状细胞病);外在因素(免疫性溶血、感染、毒物、药物、微血管病性溶血、脾功能亢进)。
- 失血性:急性大出血与慢性失血(消化道失血、月经过多)。
发病机制
- 生成不足:铁、叶酸、维生素B12等原料缺乏或骨髓造血受抑/克隆异常,导致网织红细胞生成低下。
- 溶血:红细胞寿命缩短(血管内/外溶血),胆红素升高、网织红细胞代偿性增多。
- 失血:循环血容量与红细胞总量减少,慢性失血可致铁耗竭。
- 人体的铁代谢
| 部分 | 要点 | 简明说明 |
|---|---|---|
| 体内分布 | 大部分铁以血红素形式存在于红细胞内 | 成人体内总铁约3–4 g:约65–70%存在于血红蛋白/肌红蛋白中,20–30%以储存铁(铁蛋白/含铁血黄素)形式存在于肝、脾与骨髓,其余以转铁蛋白结合于血浆或作为细胞内辅因子存在。 |
| 来源 | 膳食与内源性循环再利用 | ①膳食:动物性(heme iron,肌红蛋白/血红素铁)与植物性(non‑heme iron,多为Fe3+);②内源性:衰老红细胞破坏后释放的铁被网状内皮系统回收并再利用。 |
| 排泄 | 日常丢失量小(≈<1 mg/d) | 主要经粪便丢失(含脱落肠上皮细胞与未吸收的铁,<1 mg/日);尿中、汗液中丢失很少;哺乳期通过乳汁约1 mg/日。无专门的主动排铁途径。 |
| 吸收 | 主要在十二指肠及空肠上段吸收为Fe2+ | heme(血红素)铁可被直接吸收;non‑heme铁多为Fe3+,需先被还原为Fe2+或形成可溶性螯合物才被吸收。维生素C等还原剂促进Fe3+→Fe2+,蛋白酶解产物(某些氨基酸、肽)增加铁溶解度并促进吸收。胃酸有助于铁的溶解与还原,因此胃酸减少或抑酸剂可降低铁吸收。 |
| 运输与利用 | 血浆中以转铁蛋白转运Fe3+ | 肠上皮细胞将吸收的Fe2+释放入血后被氧化为Fe3+并结合到转铁蛋白(每分子转铁蛋白可结合2个Fe3+)。血浆转铁蛋白饱和度正常约30–35%(即约1/3被铁饱和)。转运到组织后,铁从转铁蛋白释放并还原为Fe2+,用于造血(血红蛋白合成)及细胞内铁硫蛋白、细胞色素等功能;多余铁被储存为铁蛋白/含铁血黄素。 |
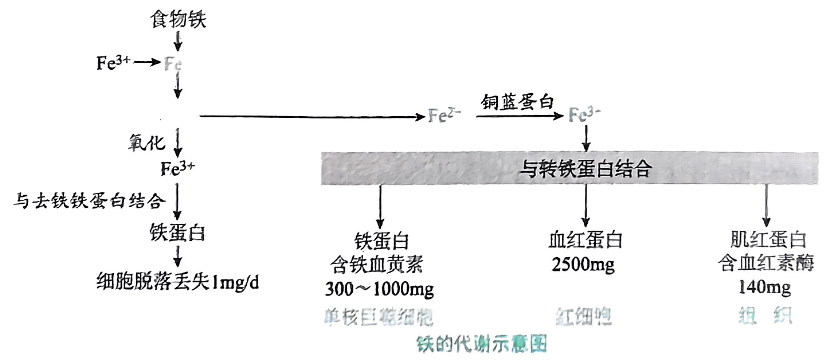
临床表现
- 一般:乏力、头晕、心悸、气促、皮肤黏膜苍白;重度可出现劳力性呼吸困难、心功能不全。
- 溶血提示:黄疸、脾大、尿色深(血红蛋白尿)。
- 营养缺乏线索:舌炎、口角炎、匙状甲;维生素B12缺乏可有神经系统症状(后索/锥体束受累)。
| 系统 | 主要表现 | 说明/临床要点 |
|---|---|---|
| 总体 | 乏力,为贫血最常见的全身症状;可伴苍白、心悸、气短。 | 按严重程度分轻中重,影响活动耐力与生活质量。 |
| 神经系统 | 头痛、眩晕、注意力不集中、记忆力下降、失眠、多梦、耳鸣、视物模糊。 | 慢性重度贫血可出现脑缺氧相关的神经症状,必要时需评估认知与情绪。 |
| 皮肤粘膜 | 皮肤和粘膜苍白,溶血性贫血可见黄疸与黏膜色素改变。 | 检查指(掌)甲、结膜、口唇及舌的苍白或黄染,有助判断贫血类型。 |
| 呼吸系统 | 活动后呼吸加深加快,重度贫血可有端坐呼吸或呼吸困难。 | 与心肺储备有关,需与肺部疾病鉴别并监测血气和氧合。 |
| 循环系统 | 心悸、心率加快、心肌缺氧表现、血压可升高;长期或严重贫血可导致缺血性心脏病或心力衰竭。 | 大量或反复输血可致铁负荷和血色素沉着,需监测心功能与铁代谢。 |
| 消化系统 | 消化功能减低、食欲下降、腹胀、排便规律及粪便性状改变,慢性失血可伴黑便或便隐血阳性。 | 查找消化道慢性失血源(如溃疡、肿瘤、痔等),必要时行胃肠镜检查。 |
| 泌尿系统 | 溶血时可见血管外溶血与血红蛋白尿或胆红素尿。 | 尿检可提示溶血性贫血;注意溶血导致的肾脏损害与胆红素代谢异常。 |
| 内分泌系统 | 长期贫血可影响甲状腺、性腺、肾上腺等腺体功能,改变激素分泌,影响红细胞生成素及胃肠激素分泌。 | 在顽固性或长期贫血患者需评估内分泌功能及红细胞生成环境。 |
| 生殖系统 | 长期贫血可致男性性功能减退,女性月经量改变(常见过多或贫血加重)。 | 评估月经史、妊娠史及性功能,寻找慢性失血或营养缺乏原因。 |
| 免疫系统 | 红细胞膜上CR1减少可影响非特异性免疫功能,反复输血可影响T细胞亚群与免疫反应。 | 输血史和溶血性贫血患者需监测传染病标志与免疫学指标。 |
| 血液系统 | 外周血细胞数、形态与生化成分发生改变,严重时骨髓造血功能受累。 | 通过外周血涂片、网织红细胞计数、骨髓检查及铁学指标明确病因与造血状态。 |
辅助检查
- 血常规与红细胞指数:平均红细胞体积(MCV)、平均红细胞血红蛋白(MCH)、平均红细胞血红蛋白浓度(MCHC)。
- 小细胞低色素(MCV<80 fL):多见于缺铁性贫血、地中海贫血。
- 大细胞(MCV>100 fL):多见于巨幼细胞性贫血、骨髓增生异常性肿瘤。
- 正细胞:急性失血或溶血。
- 分类和分度小结✨
| 分类 | MCV (fL) | MCH (pg) | 常见病因 / 特征性表现 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 大细胞性贫血 | >100 | >36 | 网织红细胞大量减少或巨幼细胞形态:巨幼细胞贫血、骨髓增生异常综合征、肝病、叶酸或维生素B12缺乏导致的恶性贫血。 | ||||||||
| 正常细胞性贫血 | 80–100 | 32–36 | 红细胞大小正常:再生障碍性贫血、纯红细胞再生障碍、溶血性贫血、急性失血后、骨髓病变相关贫血。 | ||||||||
| 小细胞低色素性贫血 | <80 | <32 | 低色素、细小红细胞:缺铁性贫血、地中海贫血(珠蛋白生成障碍)、慢性病相关贫血、铁粒幼细胞性贫血(部分)与海洋性贫血等。 | ||||||||
| 血红蛋白(g/L)分度 |
| ||||||||||
-
网织红细胞:溶血↑,生成不足↓。
-
铁代谢:“三低三高”,即血清铁、血清铁蛋白、转铁蛋白饱和度均降低;总铁结合力、血清可溶性转铁蛋白受体(sTfR)、红细胞游离原卟啉升高。骨髓铁染色(缺铁诊断金标准)。
-
溶血证据:间接胆红素↑、乳酸脱氢酶↑、结合珠蛋白↓、外周血形态(裂体、球形红细胞)。
-
维生素B12/叶酸水平;甲基丙二酸与同型半胱氨酸(维生素B12缺乏时二者均↑,叶酸缺乏仅同型半胱氨酸↑)。
诊断和鉴别诊断
- 按MCV初步分型并结合网织红细胞、铁代谢、溶血指标。
- 小细胞低色素:鉴别缺铁性贫血与地中海贫血(家族史、血红蛋白电泳)。
- 大细胞:鉴别叶酸/维生素B12缺乏与骨髓增生异常性肿瘤(骨髓巨幼样变化与原始细胞比例、染色体)。
- 正细胞:区分急性失血与溶血(溶血实验室证据)。
治疗
- 缺铁性贫血:口服二价铁剂为首选(疗程至少3个月至铁储备恢复);必要时静脉铁。口服铁剂后,先是外周血网织红细胞增多,5~10天达高峰,2周后血红蛋白浓度开始升高,2个月左右恢复正常血红蛋白正常后,仍需服用铁剂4~6个月,待铁蛋白正常后停药。
- 巨幼细胞性贫血:有神经症状提示维生素B12缺乏时先补维生素B12;叶酸缺乏补充叶酸。
- 溶血:去除诱因,免疫性溶血用糖皮质激素为首选;难治加用免疫抑制剂、利妥昔单抗或脾切除;G6PD缺乏避免氧化性药物/食物。
再生障碍性贫血
病因
- 先天性/遗传性
- 范可尼贫血、先天性角化不良、Shwachman–Diamond综合征等,常伴发育畸形、内分泌异常及肿瘤易感。
- 获得性(最常见)
- 特发性:多数为免疫介导性造血干细胞损伤,找不到明确诱因。
- 物理化学因素:苯及其同系物、有机溶剂、放射线(电离辐射)。
- 药物:氯霉素、磺胺类、抗甲状腺药、金制剂、抗肿瘤药物等。
- 病毒/感染:非A–E型肝炎相关再障、EB病毒、细小病毒B19、HIV等。
- 内分泌与生理状态:妊娠相关再障(分娩后可缓解)。
- 自身免疫及克隆性疾病相关:与阵发性睡眠性血红蛋白尿关联克隆可并存。
发病机制
- 造血干/祖细胞数量显著减少与功能受抑:T细胞异常活化,分泌干扰素-γ、肿瘤坏死因子-α等,免疫介导性抑制造血。
- 骨髓微环境异常与细胞因子网络紊乱,端粒缩短、DNA修复缺陷(遗传性再障)。
- 骨髓组织学:骨髓低增生、脂肪化明显,巨核细胞显著减少或缺如。
临床表现
- 三系减少相关症状
- 贫血:乏力、心悸、气短、面色苍白。
- 出血:皮肤黏膜出血(瘀点、鼻衄、牙龈出血、月经过多)。
- 感染:中性粒细胞减少导致发热、反复或严重感染。
- 体征
- 肝、脾、淋巴结一般不肿大(与白血病、脾功能亢进的鉴别要点);先天性病例可伴发育畸形。
- 经过
- 轻型起病隐匿,重型/极重型起病急骤,出血与感染危及生命。
辅助检查
- 外周血象:全血细胞减少;网织红细胞低(通常<1%或绝对值明显下降);中性粒细胞与血小板常显著降低;红细胞指数多为正细胞正色素或轻度大细胞。
- 骨髓:骨髓穿刺增生低下甚至“干抽”,活检见细胞性明显下降、脂肪化,巨核细胞显著减少;铁贮存常正常或增多。
- 免疫学/克隆学:可检出GPI锚定蛋白(如CD55/CD59)缺失的PNH克隆(流式细胞术);排除MDS相关染色体异常。
- 排除性检查:维生素B12/叶酸水平、肝肾功能、病毒学检测等以鉴别巨幼细胞性贫血、慢性病性贫血等。
诊断和鉴别诊断
- 诊断依据
- 持续性全血细胞减少、网织红细胞低,骨髓活检示低增生且无浸润或纤维化等继发原因。
- ✨重型/极重型再障(Camitta标准,考点)
- 骨髓细胞性:<25%,或25%–50%但造血细胞<30%;
- 满足下列至少两项:绝对中性粒细胞计数<0.5×109/L,血小板<20×109/L,网织红细胞绝对值<15×109/L(或<1%)。
- 极重型:绝对中性粒细胞计数<0.2×10^9/L。
| 类别 | 要点 | 简明说明/判断标准 |
|---|---|---|
| 一般诊断标准 | 全血细胞减少 | 网织红细胞百分比<1%,出现全系或多系细胞减少。排除其他可致全血细胞减少的疾病(如PNH、Fanconi贫血、Evans综合征、免疫相关性全血细胞减少等)。常伴肝脾通常不大或无肿大。 |
| 一般诊断标准 | 外周象与骨髓变化 | 周围血:淋巴细胞比例相对增高;骨髓:造血组织减少,造血细胞显著减少或空虚。 |
| 重型再障 | 三项中至少2项或更严 | 满足下列3项中的至少2项:①网织红细胞绝对值<15×10^9/L;②中性粒细胞<0.5×10^9/L;③血小板<20×10^9/L。重型者临床常有严重感染、出血或输血依赖。 |
| 极重型提示 | 更低血象或合并严重并发症 | 如中性粒细胞极低、进行性出血或无法控制的感染、频繁输血需求,提示更危重病程需紧急处理(免疫抑制或造血干细胞移植评估)。 |
| 非重型再障 | 不满足重型标准 | 外周及骨髓改变存在但未达重型诊断阈值,临床表现较轻,可随访或根据进展决定治疗。 |
- 鉴别诊断
- 骨髓增生异常综合征:病态造血显著、染色体异常;骨髓增生可低或正常,外周血形态异常。
- 急性白血病:骨髓原始细胞增多、肝脾淋巴结肿大,外周血可见原始细胞。
- 巨幼细胞性贫血:MCV增高、骨髓巨幼样改变、维生素B12/叶酸缺乏。
- 脾功能亢进:肝脾大、网织红细胞增高、骨髓增生亢进。
- 药物/感染性骨髓抑制需结合病史及动态观察。
治疗
- 一般与支持治疗
- 去除诱因,成分输血(优选照射去白红细胞;血小板出血指征时输注),防治感染(中性粒细胞显著低下用经验性广谱抗菌),必要时G-CSF短期应用;预防铁过载(铁螯合)。
- 特异治疗
- 同胞HLA相合同种异基因造血干细胞移植为年轻重型/极重型再障的首选根治治疗。
- 无合适供者或年龄较大者:抗胸腺细胞球蛋白(ATG)+环孢素A为首选免疫抑制方案,可联合G-CSF加速恢复。
- 雄激素(如达那唑)可用于部分病例或IST失败的补充治疗。
- 预后与随访
- 关注复发与克隆性演变(MDS/PNH);长期输血者监测铁负荷并行铁螯合。
溶血性贫血
病因
- 红细胞内在异常(遗传性为主)
- 膜骨架缺陷:遗传性球形红细胞增多症、遗传性椭圆形红细胞增多症、口形红细胞增多症等。
- 酶缺陷:葡萄糖-6-磷酸脱氢酶(G6PD)缺乏、丙酮酸激酶缺乏等。
- 血红蛋白病:地中海贫血、不稳定血红蛋白病、镰状细胞病等。
- 红细胞外在因素(获得性)
- 免疫性:自身免疫性溶血性贫血(温抗体型/冷凝集型)、药物诱导性免疫溶血、新生儿溶血。
- 非免疫性:微血管病性溶血性贫血(TTP/HUS/DIC)、机械性(人造瓣膜)、感染(疟疾等)、毒物/烧伤、脾功能亢进等。
发病机制
- 共同特点:红细胞破坏增加,骨髓代偿性红系增生,网织红细胞增多。
- 溶血部位
- 血管外溶血(脾/肝巨噬系统):红细胞被识别吞噬,间接胆红素升高、脾大,尿胆原↑,通常无血红蛋白尿。
- 血管内溶血(循环内破坏):血浆游离血红蛋白↑、结合珠蛋白↓、血红蛋白尿/含铁血黄素尿,乳酸脱氢酶(LDH)显著升高,肾损伤风险高。
临床表现
- 典型三联征:贫血、黄疸、脾肿大;活动期可出现发热、深色尿。
- 特殊线索
- G6PD缺乏:感染、氧化性药物或蚕豆诱发急性溶血。
- 温抗体型AIHA:常为血管外溶血,库姆斯试验阳性。
- 冷凝集型AIHA:遇寒加重,肢端紫绀/雷诺样改变,常为补体介导。
- 微血管病性:裂红细胞、血小板减少及出血/器官损害表现。
辅助检查
- 溶血证据
- 网织红细胞增多,间接胆红素↑,LDH↑,血清结合珠蛋白↓。
- 尿:尿胆原↑;血管内溶血时见血红蛋白尿或含铁血黄素尿。
- 病因线索
- 外周血涂片:球形红细胞(膜病)、靶形红细胞/小细胞(地贫)、裂红细胞(微血管病)、嗜碱性点彩等。
- 骨髓象:红系增生活跃,粒红比下降。
- 直接抗人球蛋白试验(直接库姆斯试验):免疫性溶血首选检查,阳性支持诊断。
- G6PD/丙酮酸激酶活性测定:G6PD需在非急性溶血期检测更准确。
- 红细胞渗透脆性试验/EMA结合试验:提示遗传性球形红细胞增多症。
- 血红蛋白电泳/基因分析:诊断血红蛋白病与地中海贫血。
- 流式细胞术(CD55/CD59或FLAER):筛查PNH克隆。
诊断和鉴别诊断
- 诊断要点
- 溶血实验室证据 + 红系代偿增生 + 明确病因线索。
- 免疫性溶血性贫血:直接库姆斯试验阳性(温抗体IgG为主,冷凝集IgM/补体为主)。
- 鉴别诊断
- 血管内 vs 血管外溶血(是否血红蛋白尿、结合珠蛋白显著下降、LDH显著升高)。
- 免疫性 vs 非免疫性(库姆斯试验)。
- 与单纯肝胆疾病(黄疸为主而无网织红细胞增多)及Gilbert综合征鉴别。
- 与再生障碍性贫血(网织红细胞低、骨髓低增生、无黄疸)鉴别。
治疗
- 一般治疗
- 去除诱因;补充叶酸;溶血活动期充分水化与碱化尿,预防肾损伤;必要时输注洗涤红细胞。
- 免疫性溶血性贫血
- 糖皮质激素为首选(如泼尼松约1 mg/kg·d),缓解后逐渐减量。
- 激素无效或复发:利妥昔单抗或脾切除(温抗体型);难治者可用环孢素A、硫唑嘌呤、环磷酰胺等;重症可短期静脉丙种球蛋白。
- 冷凝集病:避免寒冷,利妥昔单抗为主要药物;补体抑制策略在部分病例有效。
- 非免疫性溶血
- G6PD缺乏:严格避免氧化性诱因,发作期支持治疗。
- 遗传性球形红细胞增多症:中重度溶血或并发胆石症者行脾切除(患儿宜>5岁后择期)。
- 微血管病性溶血:按原病因处理(如TTP行血浆置换,DIC纠正凝血紊乱,HUS支持/特异治疗)。
- PNH:对溶血明显者可用补体抑制剂,合并血栓风险者重视抗凝与并发症处理。
白细胞减少
病因
- 感染:病毒性最常见(上呼吸道病毒、肝炎病毒等)。
- 药物/毒物:抗甲状腺药、氯氮平、磺胺类、氯霉素、化疗药物等。
- 血液病:再生障碍性贫血、骨髓增生异常性肿瘤、白血病等。
- 自身免疫性疾病、脾功能亢进、营养不良等。
发病机制
- 生成减少(骨髓抑制/受侵)、外周破坏增加或边缘池再分布。
临床表现
- 多无特异症状;中性粒细胞绝对计数(ANC)下降时感染风险显著增加,<1.0×109/L为高危,<0.5×109/L为极高危。
辅助检查
- 血常规三系;外周血涂片;必要时骨髓检查。
- 病因学筛查(药物史、感染标志物、自免相关抗体/甲状腺功能等)。
诊断与治疗
- 明确病因,停可疑致病药物;轻中度可观察。
- 感染高危或伴发热者:经验性广谱抗菌治疗。
- 促白细胞生成素(G‑CSF)用于中重度粒细胞减少或化疗后。
- 持续/不明原因须骨髓病变排查。
粒细胞缺乏症
病因
- 多与药物相关(抗甲状腺药、氯氮平、磺胺类、氯霉素、化疗药)。
- 自身免疫、放射/毒物、严重感染等。
发病机制
- 粒细胞极度减少(通常指ANC<0.5×109/L,重型<0.2×109/L),感染控制能力丧失。
临床表现
- 起病急、高热、口腔/咽部坏死性溃疡、肛周感染,迅速进展为败血症与休克;最常见死因为严重感染/脓毒症。
辅助检查
- 血常规显著中性粒细胞减少或几乎为0;培养寻找病原体;必要时骨髓检查(药物免疫性多为成熟粒细胞缺如)。
诊断和鉴别诊断
- 与一般白细胞减少按严重程度与起病速度鉴别;排除白血病/再障/骨髓增生异常性肿瘤。
治疗
- 立即停致病药;尽早经验性广谱抗生素(需覆盖铜绿假单胞菌等革兰阴性菌,并兼顾革兰阳性球菌)。
- G‑CSF、严格防护隔离、局部创面处理;难治或感染危重可考虑粒细胞输注。
- 并发脓毒性休克按指南复苏与器官支持。
骨髓增生异常性肿瘤/⻣髓增⽣异常综合征 (MDS)
病因
- 克隆性造血干细胞疾病,部分与放化疗/苯暴露相关;年龄增高是重要危险因素;常见体细胞突变(如SF3B1、TET2、ASXL1等)。
发病机制
- 无效造血与血细胞形态学异常,多系细胞减少;可进展为急性髓系白血病(“转白”)。
临床表现
- 以贫血为主,伴感染/出血倾向;部分有脾大。
辅助检查
- 外周血:单/双/三系减少,MCV常偏大。
- 骨髓:一般为增生活跃但无效造血,原始细胞<20%;可见铁粒幼细胞增多。
- 细胞遗传学:常见−5/5q−、−7/7q−、+8、20q−等。
- 风险分层:IPSS‑R(细胞遗传学、血细胞计数、原始细胞比例)。
诊断和鉴别诊断
- 满足持续性细胞减少、骨髓发育异常与/或克隆学异常,排除维生素B12/叶酸缺乏、再障、药物性抑制等。
- FAB分型:难治性贫血(RA)、环形铁粒幼细胞性难治性贫血(RAS)、难治性贫血伴原始细胞增多(RAEB)、难治性贫血伴原始细胞增多转变型(RAEB-t)、慢性粒-单核细胞性白血病(CMML)
| FAB分型 | 外周血(要点) | 骨髓(要点) |
|---|---|---|
| RA | 原始细胞 <1% | 原始细胞 <5% |
| RAS | 原始细胞 <1% | 原始细胞 <5%,环形或不成熟粒细胞(含有核红细胞)约15% |
| RAEB | 原始细胞 <5% | 原始细胞 5%–20% |
| RAEB‑T / RAEB‑I | 原始细胞 ≥5% | 原始细胞 >20% 且 <30%;或出现幼粒细胞/Auer小体 |
| CMML | 原始细胞 <5%,单核细胞增多>1×10^9/L | 原始细胞 5%–20% |
治疗
- 低危:支持治疗(促红素±G‑CSF)、来那度胺(5q‑特异获益)、铁螯合(铁过载)。
- 中高危:去甲基化药物(阿扎胞苷/地西他滨);适合者同种异体造血干细胞移植为唯一根治手段。
- 感染/出血对症处理。
急性白血病
分子病理和治疗很重要!
病因
- 遗传易感、辐射/化学暴露、病毒、既往化疗等;关键分子事件如t(15;17)、t(8;21)、inv(16)、BCR‑ABL等。
发病机制
- 白血病克隆异常增殖并抑制正常造血,引起贫血、出血、感染三大主症;浸润肝脾淋巴结、中枢神经等。
临床表现
- 乏力、发热、出血点瘀斑,肝脾淋巴结肿大、骨痛;APL(急性早幼粒细胞白血病)易并发DIC。
| 症状 | 临床特点 | 备注 |
|---|---|---|
| 贫血 | 部分病人病程短,可无明显贫血;多数就诊时已出现不同程度贫血 | 半数病人就诊时已有严重贫血,尤以继发MDS者明显 |
| 发热 | 多以发热为早期表现,可有低热或高热,高热提示继发感染 | 最常见感染部位:口腔、牙龈、咽喉;常见致病菌含革兰氏阴性杆菌、肺炎克雷伯菌、铜绿假单胞菌、大肠埃希菌等 |
| 出血 | 可见皮肤淤点、瘀斑、鼻出血、牙龈出血、月经过多 | 急性早幼粒细胞白血病易并发弥散性血管内凝血;急性白血病死于出血者约62%,其中87%为颅内出血 |
| 淋巴结肿大 | 多见于ALL | 纵隔淋巴结肿大多见于T细胞急性淋巴细胞白血病(T-ALL) |
| 肝、脾大 | 肝、脾多为轻至中度肿大 | 巨脾常见于慢性髓系白血病或白血病性变 |
| 骨骼与关节 | 常有胸骨下段或局部压痛,发生骨髓坏死可引起剧痛 | 可出现关节痛、骨痛,儿童多见 |
| 眼部 | 粒细胞肉瘤(绿色瘤)常累及巩膜,以眼眶累及常见 | 多见于急性髓细胞白血病(AML) |
| 口腔 | 白血病细胞浸润可导致牙龈增生、肿胀、出血 | 多见于急性髓-单(M4)和单(M5)亚型 |
| 皮肤 | 可出现蓝灰色或红褐色结节、隆起、变硬或蓝色斑节 | 多见于急性髓-单(M4)和单(M5)亚型 |
| CNSL(中枢神经系统浸润) | 最常见的颅内浸润病灶;化疗药物难以通过血脑屏障 | 多见于ALL化疗缓解期儿或成人,需鞘内或全身强化治疗 |
| 睾丸 | 次常见的体外浸润部位,常为单侧、无痛性肿大 | 多见于ALL化疗缓解后的幼儿和青年男性 |
分类
- 急性髓系白血病(AML)的分型:
红9为1,红3为2,早起为3; 原粒;原始;原始单核
| 中文名 | 特点 |
|---|---|
| M0 急性髓细胞性白血病微分化型 | 原始细胞>30%,无嗜天青颗粒及Auer小体;髓过氧化物酶(MPO)及苏丹黑B阳性细胞<3%;MPO可阴性;髓系抗原(如CD33或CD13)可呈阳性,淋系抗原通常为阴性;血小板抗原可阴性 |
| M1 急性粒细胞白血病未分化型 | 原粒细胞占骨髓非红系有核细胞(NEC)>90%,MPO阳性细胞>3% |
| M2 急性粒细胞白血病部分分化型 | 原粒细胞占骨髓NEC 30%~89%,其他粒细胞≥10%,单核细胞<20% |
| M3 急性早幼粒细胞白血病 | 骨髓以颗粒增多的早幼粒细胞为主,早幼粒细胞在NEC中占比≥30% |
| M4 急性粒–单核细胞白血病 | 骨髓原始细胞及NEC>30%,各阶段粒细胞≥20%,各阶段单核细胞≥20% |
| M5 急性单核细胞白血病 | 骨髓NEC中原始单核细胞、幼稚单核细胞>30%,且原始单核细胞、幼稚单核细胞及单核细胞≥80% |
| M6 红白血病 | 骨髓中幼红细胞>50%,NEC中原始细胞≥30% |
| M7 急性巨核细胞白血病 | 骨髓中原始巨核细胞>30%,血小板抗原可阳性,血小板过氧化酶阴性 |
- 急性淋巴细胞白血病(ALL)的分型
| 分型 | 病理特点 |
|---|---|
| L1 | 以小细胞为主(直径≤12μm),细胞大小均一,核质比低,核仁不明显,胞质少。 |
| L2 | 以大细胞为主(直径>12μm),形态多样,染色质较粗,核仁可见,细胞异型性增加。 |
| L3(Burkitt型) | ✨细胞较大且大小一致,胞内有明显空泡,胞质嗜碱性,染色深,核分裂像多,提示Burkitt样高度增殖特征。 |
辅助检查
- 血常规白细胞可高可低;外周/骨髓涂片见原始/幼稚细胞。
- 骨髓原始细胞比例:≥20%支持急性白血病(特定遗传学异常可<20%亦诊断)。
- 免疫分型、细胞遗传学与分子学(风险分层与靶向治疗依据)。
P亮M藏为淋,NSE过半为单。单粒有小体。
| 染色/标志 | 急性淋巴细胞白血病 (ALL) | 急性粒细胞白血病 (AML) | 急性单核细胞白血病 |
|---|---|---|---|
| MPO | 阴性 (-) | 分化差的原始/杆状细胞 阴性~弱阳性;分化好者 阳性~强阳性 | 可为 阴性~弱阳性 |
| PAS | 阳性:成块或粗颗粒状 | 可为(-)或(+);常见弥漫性淡红色或细颗粒状 | 可为(-)或(+);常见弥漫性淡红色或细颗粒状 |
| NSE (α-萘酚-ASD-苯胺苷酶) | 通常阴性 (-) | (-)或(+),NaF 抑制率 <50% | 阳性 (+),NaF 抑制率 ≥50% |
| NAP (中性粒细胞碱性磷酸酶) | 增加 | 减少或接近阴性 (-) | 正常或减低 |
| Auer 小体 | 无 (-) | 可见 (+) | 可见 (+) |
- 特征:AML常见Auer小体;CML有BCR‑ABL融合基因。
诊断和鉴别诊断
- 急性髓系白血病与急性淋巴细胞白血病鉴别依赖免疫表型与分子学。
- 与反应性白细胞增多、MDS、再障鉴别。
治疗
急髓A,急淋V(
谁临AV),M3全反加砒霜。
- AML诱导:蒽环类+阿糖胞苷(“DA方案”),
一般有A(阿糖胞苷)的2个字母的就对了;巩固/强化±造血干细胞移植。 - APL:全反式维甲酸(ATRA) ± 三氧化二砷为首选,并积极防治DIC。
- ALL:联合化疗(常用VDLP方案),
一般有V(长春新碱)就对了。中枢神经系统预防(鞘内化疗/颅放疗)。
| 疾病 / 适应 | 诱导/方案 | 主要药物与要点 |
|---|---|---|
| 急性淋巴细胞白血病 (ALL) | VP(基础方案) | VCR(长春新碱) + P(泼尼松或泼尼松龙);作为基本化疗方案 |
| DVP(成人常用) | DNR(柔红霉素) + VCR(长春新碱) + P(泼尼松);成人常用方案 | |
| DVLP(最常用) | DNR + VCR + L-ASP(左旋门冬酰胺酶) + P;常用且疗效良好 | |
| 急性髓系白血病 (AML, 非早幼粒型) | IA | IDA(伊达比星) + Ara‑C(阿糖胞苷);非APL常用诱导方案 |
| DA | DNR(柔红霉素) + Ara‑C;非APL常用方案 | |
| HA(已少用) | HHT(高三尖杉酯碱) + Ara‑C;过去常用、现已弃用 | |
| 急性早幼粒细胞白血病 (APL, M3) | ATRA + 蒽环类(诱导首选) | 首选全反式维A酸 (ATRA) 联合蒽环类药物;ATRA 能显著提高完全缓解率(≈85%),并需同时注意凝血障碍的支持治疗(输血小板、新鲜冰冻血浆等) |
| ATRA + 蒽环类 + ATO(联合) | 加入ATO(砷剂)可缩短达到完全缓解的时间,适用于多数APL患者 | |
| ATRA + ATO(双诱导) | 适用于低/中危或不能耐受蒽环类者;为非化疗或减化疗策略 | |
| 辅助/支持治疗 | 预防并处理弥散性凝血功能障碍(DIC):及时输注血小板/新鲜冰冻血浆、监测凝血指标;诱导期警惕分化综合征并给予类固醇处理 |
慢性髓系白血病
病因
- 起源于造血干细胞的克隆性恶性增殖性疾病,属于骨髓增殖性肿瘤的一型。
- 关键分子事件为染色体易位 t(9;22)(q34;q11),形成费城染色体,产生 BCR‑ABL1 融合基因,编码持续活化的酪氨酸激酶,是本病的特征性分子标志(诊断关键)。
- BCR‑ABL1不同转录本:以p210型(b2a2/b3a2)最常见并典型;p190多见于淋巴系白血病;p230较少见。
- 危险因素:电离辐射、苯及其同系物等,但多数患者无明确暴露史。
发病机制
- BCR‑ABL1酪氨酸激酶持续激活下游信号(RAS/MAPK、PI3K/AKT、JAK/STAT),导致过度增殖、凋亡减少、黏附下降,大量髓系祖细胞释放入外周。
- 出现显著的粒系增殖和成熟谱齐全的“左移”(从中幼粒细胞至成熟中性粒细胞),并常伴嗜碱性粒细胞、嗜酸性粒细胞增加。
- 代谢活性增强导致尿酸增高、LDH升高;髓外造血引起脾脏明显肿大。
- 自然病程分三期:慢性期→加速期→急变期(急变期相当于急性白血病)。
临床表现
- 起病隐匿,常以乏力、体重下降、夜间盗汗、腹胀(左上腹)就诊。
- 体征以脾大(常中—重度)最具提示性,可达脐下;肝亦可肿大。
- 高白细胞相关症状:头痛、视物模糊、气促、甚至白细胞滞留综合征(特别是白细胞极高时)。
- 其他:痛风/高尿酸血症,轻—中度贫血;血小板可高可低(功能异常可致出血倾向)。
- 进入加速/急变期的警讯:发热、骨痛、脾迅速增大,外周原始细胞比例上升。
辅助检查
- 外周血:
- 白细胞显著增高,常>50×10^9/L,粒系谱齐全左移,可见晚幼粒—中幼粒等;嗜碱性粒细胞比例升高。
- 血小板增高较常见,晚期可减少;贫血多轻—中度。
- 中性粒细胞碱性磷酸酶(NAP)积分低(与类白反应鉴别的重要指标)。
- 骨髓象:增生活跃至极度活跃,粒系显著增生,粒/红比高(常>10:1),巨核细胞增多(可见小型巨核细胞),进展期可见纤维化加重。
- 细胞遗传学/分子:
- 费城染色体阳性;BCR‑ABL1融合基因阳性(FISH/定量PCR)为确诊依据。
- 治疗监测以BCR‑ABL1定量PCR(国际标准化比值)为主。
- 生化:尿酸、LDH升高常见;肝肾功能据伴随情况改变。
- 影像:脾脏增大(超声/CT评估)。
诊断和鉴别诊断
- 诊断要点:
- 典型临床与血象(白细胞显著增高、谱齐全左移、脾大);
- 骨髓粒系增生为主;
- 费城染色体或BCR‑ABL1融合基因阳性(金标准)。
- 分期(要点化记忆):
- 慢性期:原始细胞<10%。
- 加速期:原始细胞10%–19%,或嗜碱性粒细胞≥20%,或血小板持续异常(<100或>1000×10^9/L且难以纠正),或克隆演变等。
- 急变期:外周/骨髓原始细胞≥20%,或出现髓外原始细胞浸润。
- 常见鉴别:类白血病反应、慢性粒‑单核细胞白血病(CMML)、非典型慢性髓系白血病(aCML)、其他骨髓增殖性肿瘤(真性红细胞增多症、原发性血小板增多症、原发性骨髓纤维化)等。见下表要点比较。
| 要点 | 慢性髓系白血病 | 类白血病反应 | 慢性粒‑单核细胞白血病/非典型慢性髓系白血病 |
|---|---|---|---|
| 病因标志 | BCR‑ABL1阳性(费城染色体) | BCR‑ABL1阴性,多继发感染/肿瘤等刺激 | 多为BCR‑ABL1阴性;可见其他克隆异常(如SETBP1等突变,实验室层面需专门测序) |
| 白细胞 | 显著增高,粒系谱齐全左移,嗜碱性↑ | 中/重度增高可见,谱亦可左移,嗜碱性不显著↑ | 白细胞↑,单核细胞持续≥1×10^9/L且≥10%(CMML);aCML以失调性改变明显 |
| 脾脏 | 明显肿大常见 | 一般不大或轻度大 | 可肿大,但通常较CML轻 |
| NAP积分 | 低 | 高或正常 | 多为正常或偏低,需结合其他指标 |
| 骨髓象 | 粒系显著增生,巨核细胞↑(可见小型巨核) | 反应性改变,感染控制后可恢复 | 可见失调性改变、单核细胞系增生(CMML) |
| 其他实验室 | 尿酸、LDH↑ | 随原发病改变 | CMML常有细胞遗传学/分子异常,贫血、血小板异常较常见 |
| 结论 | 检出BCR‑ABL1即确诊 | 去除诱因后自行恢复 | 需分子学/骨髓病理综合判定,BCR‑ABL1阴性支持其诊断 |
治疗
- 总则与目标
- 首选:酪氨酸激酶抑制剂。目标为快速达成并维持深度分子学缓解,降低进展为加速/急变期的风险。
- 里程碑监测(BCR‑ABL1国际标准化比值,外周血定量PCR):3个月≤10%、6个月≤1%、12个月≤0.1%(主要分子学缓解,MMR)。未达标需评估依从性、药物相互作用并及时调整方案。
- 初治慢性期
- 一线药物:伊马替尼;亦可选择达沙替尼、尼洛替尼(高危或需更快深度缓解者常用)。
- 白细胞极高伴症状时:可短期用羟基脲降白或行白细胞去除术以防白细胞滞留;同时尽快启动酪氨酸激酶抑制剂。
- 支持治疗:别嘌醇/静脉补液预防高尿酸并发症;根据需要纠正贫血、出血与感染。
- 耐药或不耐受
- 更换其他代次的酪氨酸激酶抑制剂,并行BCR‑ABL1激酶结构域突变检测以指导选择;T315I突变对多数药物耐药,优选具有相应活性的药物或考虑移植。
- 加速/急变期
- 选用更强效的酪氨酸激酶抑制剂联合相应化疗(急变为髓系/淋巴系分别按方案处理),同种异基因造血干细胞移植为根治手段,尤其对耐药或高危者。
- 停药尝试
- 达到并维持深度分子学缓解(如MR4或更深)≥1–2年、严格监测条件下,部分患者可尝试停药以实现“治疗后无治疗缓解”。停药后需高频次PCR监测,分子学复发应迅速恢复治疗。
- 不良反应与个体化选择(考点提示)
- 伊马替尼:水肿、肌痉挛、皮疹、胃肠道反应、肝酶升高。
- 达沙替尼:胸腔积液、肺动脉高压风险。
- 尼洛替尼:代谢异常、动脉血栓事件风险,注意心血管评估。
- 泊那替尼:血栓与血管事件风险高,多用于多线耐药或特定突变。
- 妊娠期:优选干扰素‑α;避免酪氨酸激酶抑制剂与羟基脲。
霍奇金淋巴瘤
病因
- 病毒(EB病毒等)、免疫缺陷/免疫紊乱、环境因素等;本质为淋巴细胞克隆性肿瘤。
发病机制
- 霍奇金淋巴瘤(HL)与非霍奇金淋巴瘤(NHL)生物学差异显著。
临床表现
- 无痛性、进行性淋巴结肿大(颈部常见),B症状(发热、盗汗、体重下降),脾肿大;HL常呈连续性向邻近淋巴区转移并可见Reed‑Sternberg细胞。
辅助检查
- 组织学活检(切除活检首选);免疫表型、分子学;影像分期(PET‑CT)。
分组与分期
Ann Arbor 分期系统(1989)
| 项目 | 分类/期别 | 要点(简明) |
|---|---|---|
| 全身症状分组 | A 组 / B 组 | 若无以下任一全身症状为A组;若出现下列任一者为B组:① 不明原因发热>38℃;② 盗汗(夜间大量出汗);③ 半年内体重下降≥10%。 |
| I 期 | 局限单侧单区 | 单个淋巴结区域(I)或局灶性单个结外器官(IE)受侵犯,局限性病变,系统影响少。 |
| II 期 | 同侧多区或伴局灶结外累及 | 同侧淋巴结两组或多组受侵犯(II),或局灶性单个结外器官及其邻近淋巴结受侵(IIE);可伴或不伴横膈膜另一侧淋巴结受侵犯。 |
| III 期 | 隔膜两侧累及 | 横膈上、下淋巴结同时受累(III),可伴局灶性相关结外器官(IIIE)、脾受侵犯(IIIS)或二者兼有(IIIS+E)。 |
| IV 期 | 弥漫或多发性结外累及 | 弥漫性(多灶性)单个或多个结外器官受侵犯,伴或不伴相关淋巴结肿大,或孤立性结外器官受侵并有远处(非区域性)淋巴结肿大。若肝或骨髓受累,或出现器官功能受损,均属IV期。 |
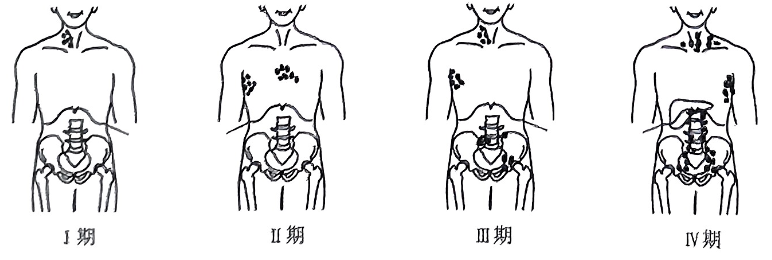
治疗
- HL:ABVD=A(多柔比星)+B(博来霉素)+V(长春碱)+D(达卡巴嗪)±放疗。
非霍奇金淋巴瘤
病因
- 感染相关
- 幽门螺杆菌与胃黏膜相关边缘区淋巴瘤(胃MALT)密切相关,根除感染可诱导缓解。
- EB病毒:与内鼻型NK/T细胞淋巴瘤、部分伯基特淋巴瘤、移植后淋巴增殖性疾病相关。
- 人类T细胞白血病/淋巴瘤病毒1型(HTLV‑1):成人T细胞白血病/淋巴瘤。
- 丙型肝炎病毒:与边缘区淋巴瘤相关;人类免疫缺陷病毒:免疫缺陷状态下NHL显著增加。
- 免疫状态与自身免疫
- 器官移植/免疫抑制、原发或继发免疫缺陷。
- 自身免疫病(干燥综合征、类风湿关节炎、乳糜泻等)增加风险。
- 遗传与分子学
- 染色体易位与基因异常:滤泡性淋巴瘤t(14;18)/BCL2过表达;伯基特淋巴瘤t(8;14)/MYC;弥漫大B细胞淋巴瘤常见BCL6、BCL2、MYC异常,双打击/三打击预后差;边缘区淋巴瘤t(11;18)/MALT1等。
- 环境与其他
- 放射、部分有机溶剂/农药暴露;年龄增长(多见于中老年)、男性略多。
发病机制
- 来源于成熟阶段不同的B细胞、T细胞或NK细胞的克隆性肿瘤,因体细胞突变、易位致抗凋亡/促增殖信号上调(如BCL2、MYC),或微环境慢性抗原刺激(如幽门螺杆菌)导致持续增殖与转化。
- B细胞淋巴瘤常伴免疫球蛋白重/轻链基因重排;T/NK细胞淋巴瘤伴T细胞受体基因重排。
- 结外淋巴瘤与局部黏膜相关淋巴组织(MALT)获得性形成和抗原驱动有关。
临床表现
- 淋巴结受累:无痛性、进行性淋巴结肿大,可呈多站受累,非连续性播散;纵隔、腹膜后常见。
- 结外受累:结外发生率高于霍奇金淋巴瘤,最常见部位为胃(胃肠道),亦可累及皮肤、口咽/扁桃体、甲状腺、骨、睾丸、中枢神经系统等,对应部位出现相应症状(消化不良、出血/梗阻、皮损、鼻衄/鼻塞、中枢神经定位体征等)。
- 全身症状:B症状(三联):不明原因发热、盗汗、半年内体重减轻≥10%;乏力、瘙痒(NHL较HL少见)。
- 器官浸润与压迫:肝脾肿大、骨髓受累致贫血/血小板减少/中性粒细胞减少;纵隔或腹膜后巨大肿块可致压迫症状。
- 实验室提示:乳酸脱氢酶(LDH)与β2微球蛋白升高提示肿瘤负荷与预后不良。
辅助检查
- 基本化验:血常规、LDH、β2微球蛋白、肝肾功能、电解质、尿酸;乙肝/丙肝/艾滋病筛查(单抗治疗前尤须评估乙肝并预防再激活)。
- 影像学:增强CT评估病灶;PET‑CT为分期与疗效评估的首选影像学手段,可检出隐匿病灶、指导活检与放疗靶区。
- 侵袭性评估:骨髓穿刺+活检(评估IV期);高危或症状提示时行腰穿评估中枢受累。
- 病理学(诊断核心)
- 整块/切除淋巴结活检为首选标本(避免仅细针穿刺),行形态学+免疫表型(免疫组化/流式)+分子遗传学(基因重排/FISH/突变)。
- 免疫表型:B细胞(CD19、CD20、PAX5等)、T细胞(CD3、CD5等);Ki‑67评估增殖;特异异常如环形细胞周期蛋白D1过表达提示套细胞淋巴瘤等。
- 分期与评分
- Ann Arbor/Lugano分期:I–IV期;“E”指邻近结外受累;“B”指有B症状;“巨大肿块(bulky)”常指最大径≥7–10 cm。
- 国际预后指数(IPI)五要素:年龄>60岁、LDH升高、体能状态差(ECOG≥2)、III–IV期、结外受累>1处;风险分层用于指导治疗与预后判断。
诊断和鉴别诊断
病理快送:病理学 - 淋巴瘤
- 诊断要点
- 依据病理形态+免疫表型+分子遗传学明确亚型;结合PET‑CT、骨髓活检分期和肿瘤负荷(LDH、β2微球蛋白)。
- 确认是否存在可逆病因(如幽门螺杆菌)与特殊高危因素(中枢/睾丸/肾上腺等部位)。
- 常见鉴别
- 霍奇金淋巴瘤、反应性淋巴结炎、结核性淋巴结炎、转移癌、慢性淋巴细胞白血病/小淋巴细胞淋巴瘤、传染性单核细胞增多症等。
| 疾病 | 特征 | 关键鉴别点 |
|---|---|---|
| 非霍奇金淋巴瘤 | 无痛性淋巴结肿大,结外受累常见(胃最常见),可有B症状 | 病理为克隆性淋巴瘤细胞;免疫表型界定B/T/NK;PET‑CT示高代谢多灶散在,非连续性播散;LDH常升高。 |
| 霍奇金淋巴瘤 | 多沿淋巴结连续性传播,纵隔受累常见,瘙痒较多 | 病理见典型Reed‑Sternberg细胞,CD15/CD30阳性、CD45阴性;结外受累少于NHL。 |
| 结核性淋巴结炎 | 颈部淋巴结肿大伴压痛,波动或瘘管 | 超声/影像中心干酪样坏死;抗酸染色/分子证据阳性;发热盗汗体重下降可与B症状相似,抗结核治疗有效。 |
| 反应性淋巴结炎 | 疼痛性或压痛性肿大,多与感染相关,自限 | 病理为反应性增生,无克隆性重排;随访缩小,对抗感染治疗反应好。 |
| 慢性淋巴细胞白血病/小淋巴细胞淋巴瘤 | 老年多见,外周血淋巴细胞↑,脾大 | 免疫表型典型(CD5、CD23阳性等);骨髓受累显著;进展缓慢。 |
| 转移癌(淋巴结) | 原发实体瘤既往史或体征提示 | 病理为上皮来源,角蛋白阳性;影像原发灶证据;治疗以原发瘤为主。 |
治疗
- 总体策略
- 依据亚型(惰性/侵袭性)、分期、肿瘤负荷与IPI决定。惰性型多以“观察等待”或轻强度治疗为主,侵袭性/高度恶性需强化联合方案。
- 重要部位(睾丸、肾/肾上腺、乳腺、骨髓、多结外、LDH显著升高)常需中枢神经系统预防(鞘内甲氨蝶呤/阿糖胞苷或静脉大剂量甲氨蝶呤)。
- 乙型肝炎既往感染或携带者接受抗CD20单抗前后需全程抗病毒预防。
- 常见亚型要点
- 弥漫大B细胞淋巴瘤(成人NHL中最常见):标准初治首选利妥昔单抗联合环磷酰胺、多柔比星、长春新碱、泼尼松方案(R‑CHOP)。局限期可3–4周期后合并局部放疗;进展期多为6周期。双/三打击或原发纵隔型可选加强方案(如剂量调整EPOCH联合利妥昔单抗)并个体化放疗。
- 滤泡性淋巴瘤(最常见惰性B细胞淋巴瘤):无症状、肿瘤负荷低者观察等待;需治疗时利妥昔单抗联合化疗(如与环磷酰胺、多柔比星、长春新碱、泼尼松或与苯达莫司汀),缓解后可行利妥昔单抗维持;局限期放疗可达长期缓解/潜在治愈。
- 胃MALT淋巴瘤:首选幽门螺杆菌根除治疗;若无感染或存在t(11;18)、根除无效者,行局部放疗或单抗联合化疗。
- 伯基特淋巴瘤:高增殖肿瘤,需强化、短程、分阶段方案并常规中枢预防(如含高剂量甲氨蝶呤/阿糖胞苷/长春新碱/环磷酰胺/蒽环类的序贯方案,联合抗CD20单抗)。
- 外周T细胞淋巴瘤:多用蒽环类联合方案(如CHOP样),初治达缓解者常自体造血干细胞移植巩固;复发可考虑靶向药或临床试验。
- 复发/难治
- 对化疗敏感的复发侵袭性淋巴瘤:大剂量化疗+自体造血干细胞移植。
- 抗CD19嵌合抗原受体T细胞治疗适用于复发/难治B细胞淋巴瘤。
- 其他靶向:如布鲁顿酪氨酸激酶抑制剂用于套细胞淋巴瘤等,按亚型选择。
- 支持治疗与并发症处理
- 肿瘤溶解综合征预防(补液、别嘌醇或拉布立酶)、感染预防、贫血/血小板减少对症处理;必要时生育力保护。
| 亚型 | 流行与特征 | 要点与首选治疗 |
|---|---|---|
| 弥漫大B细胞淋巴瘤 | 成人最常见;进展快,症状明显 | 首选R‑CHOP;局限期可合并放疗;高危部位考虑中枢预防;双/三打击需加强方案。 |
| 滤泡性淋巴瘤 | 最常见惰性;病程慢 | 观察等待;需治用利妥昔单抗联合化疗±维持;局限期放疗可长期缓解。 |
| 胃MALT | 与幽门螺杆菌相关 | 首选根除幽门螺杆菌;无效/阴性或特殊易位者行放疗/系统治疗。 |
| 伯基特淋巴瘤 | 高增殖(Ki‑67接近100%) | 强化短程序贯方案+常规中枢预防;需密切防治肿瘤溶解。 |
| 外周T细胞淋巴瘤 | 侵袭性、预后较差 | CHOP样为主;达缓解后自体移植巩固;复发考虑靶向/临床试验。 |
多发性骨髓瘤
病因
- 浆细胞克隆性增殖,分泌单克隆免疫球蛋白(M蛋白);与年龄增长、家族史、离子辐射等相关。
发病机制
- 骨髓微环境促发骨吸收增强、骨形成受抑,产生骨质破坏;轻链沉积损伤肾脏;免疫抑制致感染。
临床表现
- 高钙、肾功能不全、贫血、骨病(骨痛/病理骨折/溶骨性破坏);反复感染、体重下降。
| 受累系统 | 主要表现 | 简要机制/备注 |
|---|---|---|
| 骨骼 | 骨痛(多见于腰、骶、胸骨、肋骨) | 广泛溶骨性病变,骨髓瘤细胞破坏骨质并可见病理性骨折。 |
| 血液 | 贫血(约90%患者) | 由于红细胞生成减少(骨髓被肿瘤细胞占位、营养/促红素不足)引起。 |
| 肾 | 肾功能损害(IgD型尤为多见) | 轻链沉积/肾小管损伤、脱水、高钙血症加重,导致急慢性肾功能不全。 |
| 代谢 | 高钙血症:食欲差、恶心、乏力 | 来自广泛溶骨与肾功能下降;可致精神症状、肾损害加重。 |
| 免疫/感染 | 易感染(多克隆免疫球蛋白下降、嗜中性粒细胞减少) | 功能性免疫缺陷,反复细菌/病毒/真菌感染风险增加。 |
| 循环血液 | 高黏滞综合征:头昏、视力/听力受影响、心力衰竭表现 | 大量单克隆免疫球蛋白(尤其IgM/高浓度IgG/IgA)致血浆黏度增高。 |
| 出血 | 出血倾向:鼻出血、牙龈出血、皮肤紫癜 | 血小板减少、凝血功能异常或黏蛋白相关功能障碍所致。 |
| 系统性沉积 | 淀粉样变:舌肿、腮腺肿大、心肌肥厚、消化道症状 | 轻链淀粉样沉积(AL)可累及心、肝、肾、神经及腺体。 |
| 神经 | 神经系统损害:肌力下降、肢体麻木、感觉迟钝 | 骨髓瘤细胞直接浸润、外周神经淀粉样沉积或治疗/代谢并发症导致。 |
| 组织外浸润 | 脏器/淋巴结肿大(以肝、脾、淋巴结、肾为常见) | 局部骨髓瘤细胞浸润或继发淀粉样变引起的器官受累。 |
辅助检查
- ✨血清蛋白电泳/免疫固定:M峰;血清游离轻链κ/λ异常;尿本周蛋白(Bence‑Jones)。
- 骨髓:浆细胞≥10%支持诊断;影像(低剂量全身CT/全身MRI)示“冲孔样”溶骨。
- 骨代谢:钙↑、碱性磷酸酶多不高(区别转移瘤)。
| 检查/项目 | 要点 | 临床意义 / 处理要点 |
|---|---|---|
| 血象 | 呈进行性正细胞性贫血,红细胞在血涂片上多见“蜻蜓状/钱币状”排列;白细胞计数多正常。 | 贫血为常见表现,提示骨髓被浆细胞侵占。必要时评估输血与促红/抗感染支持。 |
| 骨髓 | 骨髓中浆细胞异常增生;免疫表型常为 CD38⁺、CD56⁺。 | 浆细胞比例及免疫表型用于判定病情与鉴别诊断。 |
| 血清M蛋白鉴定 | 血清出现M蛋白是本病的突出特征;伴随正常免疫球蛋白减少(免疫缺陷)。 | 进行免疫电泳/免疫固定以鉴定M蛋白类型并定量(作为肿瘤负荷与疗效监测指标)。 |
| 尿液检查 | 可见蛋白尿、血尿、管型尿。约90%有蛋白尿,约35%–65%患者尿中出现本周蛋白(Bence–Jones蛋白)。 | 尿轻链检测(24h尿/免疫电泳或游离轻链)对诊断及评估肾损害非常重要;出现轻链肾病需及时处理。 |
| 血钙与血磷 | 因骨质破坏可出现高钙血症;晚期肾功能不全时血磷可升高。 | 高钙需紧急处理(补液、利尿、双膦酸盐/地塞米松等);监测电解质与肾功能。 |
| 血清碱性磷酸酶(ALP) | 本病为溶骨性病变,ALP常正常或轻度升高。 | |
| 血清总蛋白/白球蛋白 | 由浆细胞分泌,与全身骨髓瘤细胞总数明显相关,可反映肿瘤负荷。 | |
| 细胞遗传学 | 荧光原位杂交(FISH)可在>90%患者发现细胞遗传学异常,常见异常:del(13q)、del(17p)、t(4;14)、t(14;16)、t(11;14)、超二倍体等。 | 染色体异常用于风险分层与预后判断(高危需强化治疗/移植评估)。 |
| 影像学检查 | X线表现:典型为圆形、边缘清楚、类似穿孔样的大小不等溶骨灶;亦可见椎体塌陷等溶骨改变。 | 常用骨系列平片、低剂量全身CT或PET-CT评估骨破坏与病灶分布,指导骨折/疼痛处理。 |
诊断和鉴别诊断
- 与华氏巨球蛋白血症、转移癌、原发性骨质疏松鉴别。
治疗
- 三联为基础:硼替佐米+来那度胺+地塞米松(具体按年龄/合并症调整),具体有:有症状骨髓瘤的初始治疗可选用硼替佐米+地塞米松(VD)、来那度胺+地塞米松(RD)、来那度胺+硼替佐米+地塞米松(VRD)、硼替佐米+多柔比星+地塞米松(PAD)等方案。
- 适合者行自体造血干细胞移植;骨病用双膦酸盐/地诺单抗;防治感染与肾损伤。
出血性疾病
病因分类
- 血小板相关:数量减少(ITP、骨髓抑制等)、功能障碍。
- 凝血因子缺乏:先天(血友病A/B、vWF病)、获得性(肝病、维生素K缺乏、口服抗凝药过量)。
- 继发性:弥散性血管内凝血(DIC)、纤溶亢进、血管壁异常等。
| 常见病因 / 机制 | 简明临床要点 | |
|---|---|---|
| 血管壁异常 | 先天性:遗传性出血性毛细血管扩张症、家族性单纯性紫癜、先天性结缔组织病;获得性:败血症、过敏性紫癜、药物性紫癜、维生素C缺乏、糖尿病、结缔组织病。 | 表现多为皮肤黏膜出血,注意纠正原发病;对有结缔组织病或坏死性病变者重视感染与血管脆弱性。 |
| 血小板异常 | 数量异常:血小板↑(原发性血小板增多症);血小板↓(破坏过多如ITP、消耗过多如DIC、生成减少如再障、脾亢)。 质量异常:遗传性(血小板无力症、巨大血小板综合征);获得性:抗血小板药物、感染、尿毒症、异常球蛋白血症引起。 | 典型表现:黏膜出血、紫癜、小点状出血。检查血小板计数与功能;疑ITP时查骨髓;DIC伴凝血因子消耗,注意处理原发病。 |
| 凝血异常 | 遗传性:血友病A/B、遗传性凝血酶原、FV、FVII、FX缺乏;获得性:维生素K缺乏、肝病导致凝血因子减少、尿毒症性凝血异常。 | 典型表现:关节内或深部出血(血友病)或手术/外伤出血异常。用PT、APTT区分外源/内源途径,补充因子或维生素K、处理肝病。 |
| 抗凝及纤溶蛋白溶解异常 | 抗凝过度:肝素、香豆素类、兔抗凝物过量、致凝剂过量;纤溶过度:免疫相关抗凝物增多、动物毒素、溶栓药物过量。 | 表现为广泛或难控出血。查药物史、凝血功能与纤溶指标(D‑二聚体、纤溶酶原活性);针对性中和(如停药、碳酸氢根、使用拮抗剂)并支持治疗。 |
| 复合性异常 | 多机制并存导致止血功能综合异常——先天性(如血管性及血友病混合)或获得性(如弥散性血管内凝血DIC)。 | 表现为同时存在血栓与出血倾向,优先识别并治疗原发病(感染、败血症、巨大创伤),并支持凝血因子/血小板输注及必要时抗纤溶/抗凝平衡处理。 |
临床特点
- 皮肤黏膜出血(瘀点/瘀斑、鼻衄、月经过多):多提示血小板问题。
- 深部出血(肌肉、关节腔出血):多提示凝血因子缺乏。
- 混合型出血:DIC、严重肝病。
| 项目 | 血管性疾病 | 血小板疾病 | 凝血障碍性疾病 |
|---|---|---|---|
| 性别 | 女性多见 | 女性多见 | 80%~90%发生于男性 |
| 阳性家族史 | 较少见 | 罕见 | 多见 |
| 出生后脐带出血 | 罕见 | 罕见 | 常见 |
| 皮肤紫癜 | 常见 | 多见 | 罕见 |
| 皮肤大片瘀斑 | 罕见 | 多见 | 可见 |
| 血肿 | 罕见 | 可见 | 常见 |
| 关节腔出血 | 罕见 | 罕见 | 多见 |
| 内脏出血 | 偶见 | 常见 | 常见 |
| 眼底出血 | 罕见 | 常见 | 少见 |
| 月经过多 | 少见 | 多见 | 少见 |
| 手术或外伤后渗血不止 | 少见 | 可见 | 多见 |
实验室模式
- ITP:血小板↓,凝血酶原时间(PT)/活化部分凝血活酶时间(APTT)正常。
- 血友病:APTT↑,PT正常,血小板正常。
- vWF病:出血时间↑,APTT可↑。
- DIC:血小板↓、PT/APTT↑、纤维蛋白原↓、D‑二聚体/FDP↑。
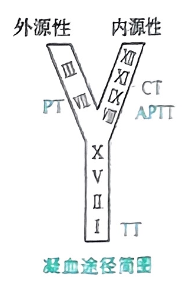
治疗
- ITP:糖皮质激素为首选;重度/危及生命:静脉丙种免疫球蛋白+应急血小板;难治可用TPO受体激动剂/利妥昔单抗/脾切除。
- 血友病:缺乏因子替代(VIII或IX);轻型A可用去氨加压素;合并出血用氨甲环酸辅助。
- vWF病:去氨加压素或vWF/FVIII浓缩物。
- DIC:去除病因;以出血为主补充血小板/新鲜冰冻血浆/冷沉淀;以血栓为主或慢性DIC可慎用肝素。
输血
基本原则
- 成分输血优先;严格“三查八对”与交叉配血;必要时紧急O型Rh阴性红细胞救治。
适应证与阈值
- 悬浮红细胞:Hb<70 g/L或有心脑缺血/进行性出血时适当放宽至<80–90 g/L;急性大出血按失血量与生命体征。
- 血小板:<10×109/L预防性;<20×109/L合并发热/感染;<50×109/L侵入性操作或活动性出血;<100×109/L重大手术/颅内出血。
- 新鲜冰冻血浆:凝血因子缺乏导致PT/APTT明显延长并出血或术前;DIC、肝功能衰竭出血。
- 冷沉淀:纤维蛋白原<1.5 g/L并出血;vWF病/血友病A在无特异制剂时。
输血不良反应与处理
- 发热非溶血反应:暂停、对症、必要时更换血制品。
- 过敏反应:抗组胺±糖皮质激素;重度过敏停输、肾上腺素。
- 急性溶血反应(最危险,常因ABO不合):立即停止输血,维持循环、利尿碱化尿、纠正DIC,保留血袋复核。
- 细菌污染性败血症:停输、广谱抗生素、循环支持。
- 输血相关急性肺损伤(TRALI):氧疗/呼吸支持,避免再用同来源浆;输血相关循环负荷过量(TACO):利尿、减慢速度。
- 迟发性溶血、同种免疫致血小板无效、移植物抗宿主病(免疫抑制患者需照射血制品)等按指南处理。